Книга RDKit ¶
Разные темы по химинформатике ¶
Д'вÅ₽Ѳㅁмаჼтичность მეფე ТАМАРА: тQönig "ICAჼ (ㅁIKB ㅐㄹ뮤") ¶
Ароматичность — одна из тех неприятных тем, которая одновременно проста и невыносимо сложна. Поскольку ни химики-экспериментаторы, ни химики-теоретики не могут прийти к согласию относительно определения, необходимо выбрать что-то произвольное и придерживаться его. Такой подход используется в RDKit.
Вместо использования шаблонов для сопоставления известных ароматических систем, код восприятия ароматичности в RDKit использует набор правил. Правила относительно просты.
Ароматичность — свойство атомов и связей в кольцах. Ароматическая связь должна быть между ароматическими атомами, но связь между ароматическими атомами не обязательно должна быть ароматической.
Например, связи слияния здесь не считаются ароматическими в RDKit:
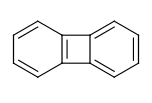
>>> from rdkit import Chem
>>> m = Chem.MolFromSmiles('C1=CC2=C(C=C1)C1=CC=CC=C21')
>>> m.GetAtomWithIdx(3).GetIsAromatic()
True
>>> m.GetAtomWithIdx(6).GetIsAromatic()
True
>>> m.GetBondBetweenAtoms(3,6).GetIsAromatic()
False
RDKit поддерживает ряд различных моделей ароматичности и позволяет пользователю определять свою собственную модель, предоставляя функцию, которая назначает ароматичность.
Модель ароматичности RDKit ¶
Кольцо или конденсированная кольцевая система считается ароматической, если она подчиняется правилу 4N+2. Вклад в число электронов определяется типом атома и окружением. Некоторые примеры:
Фрагмент |
Число пи-электронов |
c(a)a |
1 |
n(a)a |
1 |
An(a)a |
2 |
o(a)a |
2 |
s(a)a |
2 |
se(a)a |
2 |
te(a)a |
2 |
O=c(a)a |
0 |
N=c(a)a |
0 |
*(a)a |
0, 1, or 2 |
Обозначение a: любой ароматический атом; A: любой атом, включая H; *: фиктивный атом
Обратите внимание, что экзоциклические связи с электроотрицательными атомами «крадут» валентный электрон у кольцевого атома, а фиктивные атомы вносят столько электронов, сколько необходимо для того, чтобы сделать кольцо ароматическим.
Использование конденсированных колец для ароматичности может привести к ситуациям, когда отдельные кольца не являются ароматическими, но конденсированная система является таковой. Примером этого является азулен:
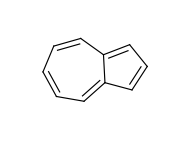
Крайний пример, демонстрирующий как конденсированные кольца, так и влияние экзоциклических двойных связей:
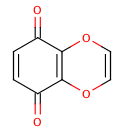
>>> m=Chem.MolFromSmiles('O=C1C=CC(=O)C2=C1OC=CO2')
>>> m.GetAtomWithIdx(6).GetIsAromatic()
True
>>> m.GetAtomWithIdx(7).GetIsAromatic()
True
>>> m.GetBondBetweenAtoms(6,7).GetIsAromatic()
False
Особый случай — гетероатомы с радикалами, которые не считаются кандидатами на ароматичность:
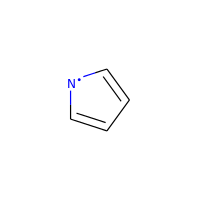
>>> m = Chem.MolFromSmiles('C1=C[N]C=C1')
>>> m.GetAtomWithIdx(0).GetIsAromatic()
False
>>> m.GetAtomWithIdx(2).GetIsAromatic()
False
>>> m.GetAtomWithIdx(2).GetNumRadicalElectrons()
1
Заряженные атомы углерода с радикалами также не рассматриваются:
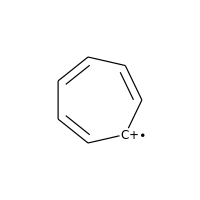
>>> m = Chem.MolFromSmiles('C1=CC=CC=C[C+]1')
>>> m.GetAtomWithIdx(0).GetIsAromatic()
False
>>> m.GetAtomWithIdx(6).GetIsAromatic()
False
>>> m.GetAtomWithIdx(6).GetFormalCharge()
1
>>> m.GetAtomWithIdx(6).GetNumRadicalElectrons()
1
Однако нейтральные атомы углерода с радикалами все еще рассматриваются:
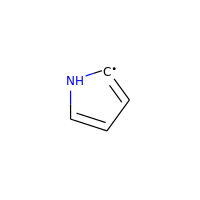
>>> m = Chem.MolFromSmiles('C1=[C]NC=C1')
>>> m.GetAtomWithIdx(0).GetIsAromatic()
True
>>> m.GetAtomWithIdx(1).GetIsAromatic()
True
>>> m.GetAtomWithIdx(1).GetNumRadicalElectrons()
1
Простая модель ароматичности ¶
Это довольно просто: только пяти- и шестичленные простые кольца считаются кандидатами на ароматичность. Используются те же самые подсчеты электронных вкладов, перечисленные выше.
Модель ароматичности MDL ¶
Это не очень хорошо документировано (по крайней мере, публично), поэтому мы попытались воспроизвести то, что указано в документации oechem ( https://docs.eyesopen.com/toolkits/python/oechemtk/aromaticity.html ).
конденсированные кольца (например, азулен) могут быть ароматическими
пятичленные кольца не являются ароматическими (хотя они могут быть частью конденсированных ароматических систем)
только C и N могут быть ароматическими
принимаются только доноры одного электрона
атомы с экзоциклическими двойными связями не являются ароматическими
Примечание: Из соображений вычислительной целесообразности восприятие ароматичности выполняется только для систем с конденсированными кольцами, где все члены имеют размер не более 24 атомов.
Поддержка и расширения SMILES ¶
RDKit охватывает все стандартные функции Daylight SMILES [ 2 ] , а также некоторые полезные расширения.
Вот (вероятно, частичный) список расширений:
Ароматичность ¶
te(ароматический Te) принимается. Вот пример с теллурофен-2-карбоновой кислотой:
>>> m = Chem.MolFromSmiles('OC(=O)c1[te]ccc1')
>>> m.GetAtomWithIdx(4).GetIsAromatic()
True
Дательные связи ¶
<-и ->создают дательную связь между атомами, направление имеет значение.
Вот пример комплекса бипиридин-медь:
>>> bipycu = Chem.MolFromSmiles('c1cccn->2c1-c1n->3cccc1.[Cu]23(Cl)Cl')
>>> bipycu.GetBondBetweenAtoms(4,12).GetBondType()
rdkit.Chem.rdchem.BondType.DATIVE
>>> Chem.MolToSmiles(bipycu)
'[Cl][Cu]1([Cl])<-[n]2ccccc2-c2cccc[n]->12'
Дативным связям присуща особая характеристика: они не влияют на валентность начального атома, но влияют на конечный атом. Так что в этом случае атомы N, участвующие в дативной связи, имеют валентность 3, которую мы ожидаем от bipy, в то время как Cu имеет валентность 4:
>>> bipycu.GetAtomWithIdx(4).GetTotalValence()
3
>>> bipycu.GetAtomWithIdx(12).GetTotalValence()
4
Кольцевые замыкания ¶
%(N)Для замыканий колец поддерживается нотация, где N — это одна цифра %(N)до пяти цифр %(NNNNN). Вот пример:
>>> m = Chem.MolFromSmiles('C%(1000)OC%(1000)')
>>> m.GetAtomWithIdx(0).IsInRing()
True
>>> m.GetAtomWithIdx(2).IsInRing()
True
Указание атомов по атомному номеру ¶
Конструкция [#6]SMARTS поддерживается в SMILES.
Четверные связи ¶
Токен $может использоваться для представления четверных облигаций в SMILES и SMARTS.
Расширения CXSMILES/CXSMARTS ¶
RDKit поддерживает синтаксический анализ и написание подмножества расширенных функций SMILES/SMARTS, представленных ChemAxon [ 4 ] .
Анализируемые функции включают в себя:
атомные координаты
()атомарные значения
$_AV:атомарные метки/псевдонимы
$(распознаваемые псевдонимы:_AP,star_e,Q_e,QH_p,AH_P,X_p,XH_p,M_p,MH_p,*)атомные свойства
atompropКоординационные/дательные связи
C(они переводятся в дательные связи)водородные связи
Hнулевой порядок облигаций облигаций
Z(пользовательское расширение, тот же синтаксис, что и c/t/ctu ниже)радикалы
^улучшенное стерео (они преобразуются в
StereoGroups)узлы ссылок
LNпеременные/многоцентровые насадки
mспецификации количества кольцевых связей
rbспецификации количества замещений без водорода
sспецификация ненасыщенности
uклиновидные связи (только при наличии атомных координат):
wU,wDволнистые облигации
wдвойная стереосвязь (только для кольцевых связей)
c,t,ctuДанные SGroup
SgDполимерные группы
SgИерархия SGroup
SgH
Функции, написанные rdkit.Chem.rdmolfiles.MolToCXSmiles()и
rdkit.Chem.rdmolfiles.MolToCXSmarts()
(обратите внимание на специализированные функции писателя), включают:
атомные координаты
атомарные значения
атомные метки
атомные свойства
дательные связки (только если дательные связки не записываются также в SMILES/SMARTS)
радикалы
улучшенное стерео
узлы ссылок
клиновидные связи (только когда также записаны атомные координаты)
волнистые облигации
двойная стереосвязь (только для кольцевых связей)
Данные SGroup
полимерные группы
Иерархия SGroup
>>> m = Chem.MolFromSmiles('OC')
>>> m.GetAtomWithIdx(0).SetProp('p1','2')
>>> m.GetAtomWithIdx(1).SetProp('p1','5')
>>> m.GetAtomWithIdx(1).SetProp('p2','A1')
>>> m.GetAtomWithIdx(0).SetProp('atomLabel','O1')
>>> m.GetAtomWithIdx(1).SetProp('atomLabel','C2')
>>> m.GetBondWithIdx(0).SetBondType(Chem.BondType.ZERO)
>>> Chem.MolToCXSmiles(m)
'C~O |$C2;O1$,atomProp:0.p1.5:0.p2.A1:1.p1.2,Z:0|'
Чтение названий молекул ¶
Если за расширениями SMILES/SMARTS и необязательными расширениями CXSMILES следуют пробел и другая строка, то анализаторы SMILES/SMARTS интерпретируют это как имя молекулы:
>>> m = Chem.MolFromSmiles('CO carbon monoxide')
>>> m.GetProp('_Name')
'carbon monoxide'
>>> m2 = Chem.MolFromSmiles('CO |$C2;O1$| carbon monoxide')
>>> m2.GetAtomWithIdx(0).GetProp('atomLabel')
'C2'
>>> m2.GetProp('_Name')
'carbon monoxide'
Эту функцию можно отключить во время анализа CXSMILES:
>>> ps = Chem.SmilesParserParams()
>>> ps.parseName = False
>>> m3 = Chem.MolFromSmiles('CO |$C2;O1$| carbon monoxide',ps)
>>> m3.HasProp('_Name')
0
>>> m3.GetAtomWithIdx(0).GetProp('atomLabel')
'C2'
Обратите внимание, что если вы отключите анализ CXSMILES, но передадите строку, содержащую CXSMILES, она будет интерпретирована как (часть) имени:
>>> ps = Chem.SmilesParserParams()
>>> ps.allowCXSMILES = False
>>> m4 = Chem.MolFromSmiles('CO |$C2;O1$| carbon monoxide',ps)
>>> m4.GetProp('_Name')
'|$C2;O1$| carbon monoxide'
Наконец, если отключить синтаксический анализ как CXSMILES, так и имен, то дополнительный текст в строке SMILES/SMARTS приведет к ошибкам: .. doctest:
>>> ps = Chem.SmilesParserParams()
>>> ps.allowCXSMILES = False
>>> ps.parseName = False
>>> m5 = Chem.MolFromSmiles('CO |$C2;O1$| carbon monoxide',ps)
>>> m5 is None
True
>>> m5 = Chem.MolFromSmiles('CO carbon monoxide',ps)
>>> m5 is None
True
Во всех примерах этого раздела использовался анализатор SMILES, но анализатор SMARTS ведет себя так же.
Поддержка и расширения SMARTS ¶
RDKit охватывает большинство стандартных функций Daylight SMARTS [ 3 ] , а также некоторые полезные расширения.
Вот (надеюсь, полный) список функций SMARTS, которые не поддерживаются:
Нететраэдрические хиральные классы
оператор
@?явные атомные массы (хотя поддерживаются запросы изотопов)
Группировка на уровне компонентов, требующая совпадений в различных компонентах, т.е.
(C).(C)
Вот (вероятно, частичный) список расширений:
Запросы гибридизации ¶
^0соответствует S гибридизированным атомам
^1соответствует гибридизированным атомам SP
^2соответствует гибридизированным атомам SP2
^3соответствует гибридизированным атомам SP3
^4соответствует гибридизированным атомам SP3D
^5соответствует гибридизированным атомам SP3D2
>> Chem.MolFromSmiles('CC=CF').GetSubstructMatches(Chem.MolFromSmarts('[^2]'))
((1,), (2,))
Дательные связи ¶
<-и ->сопоставьте соответствующие дательные связи, направление имеет значение.
>>> Chem.MolFromSmiles('C1=CC=CC=N1->[Fe]').GetSubstructMatches(Chem.MolFromSmarts('[#7]->*'))
((5, 6),)
>>> Chem.MolFromSmiles('C1=CC=CC=N1->[Fe]').GetSubstructMatches(Chem.MolFromSmarts('*<-[#7]'))
((6, 5),)
Запросы соседей гетероатома ¶
запрос атома
zсоответствует атомам, которые имеют указанное число соседей гетероатома (т.е. не C или H). Например,z2будет соответствовать второму C вCC(=O)O.запрос атома
Zсоответствует атомам, которые имеют указанное количество соседей-алифатических гетероатомов (т.е. не C или H).
>>> Chem.MolFromSmiles('O=C(O)c1nc(O)ccn1').GetSubstructMatches(Chem.MolFromSmarts('[z2]'))
((1,), (3,), (5,))
>>> Chem.MolFromSmiles('O=C(O)c1nc(O)ccn1').GetSubstructMatches(Chem.MolFromSmarts('[Z2]'))
((1,),)
>>> Chem.MolFromSmiles('O=C(O)c1nc(O)ccn1').GetSubstructMatches(Chem.MolFromSmarts('[Z1]'))
((5,),)
Запросы диапазона ¶
Диапазоны значений могут быть предоставлены для многих типов запросов, которые ожидают числовые значения. Типы запросов, которые в настоящее время поддерживают запросы диапазона:
D, h, r, R, v, x, X, z, Z, +,-
- Вот несколько примеров:
D{2-4}соответствует атомам, имеющим от 2 до 4 (включительно) явных связей.D{-3}соответствует атомам, имеющим менее или равно 3 явным связям.D{2-}соответствует атомам, имеющим не менее 2 явных связей.
>>> Chem.MolFromSmiles('CC(=O)OC').GetSubstructMatches(Chem.MolFromSmarts('[z{1-}]'))
((1,), (4,))
>>> Chem.MolFromSmiles('CC(=O)OC').GetSubstructMatches(Chem.MolFromSmarts('[D{2-3}]'))
((1,), (3,))
>>> Chem.MolFromSmiles('CC(=O)OC.C').GetSubstructMatches(Chem.MolFromSmarts('[D{-2}]'))
((0,), (2,), (3,), (4,), (5,))
Справочная информация SMARTS ¶
Обратите внимание , что текстовые версии таблиц ниже включают некоторые символы обратной косой черты для экранирования специальных символов. Это недостаток используемой нами системы документирования. Пожалуйста, игнорируйте эти символы.
Атомы ¶
Примитивный |
Свойство |
Значение по умолчанию |
Диапазон? |
НОтЫ |
|---|---|---|---|---|
a |
“aromatic atom” |
|||
A |
“aliphatic atom” |
|||
d |
“non-hydrogen degree” |
1 |
Y |
extension |
D |
“explicit degree” |
1 |
Y |
|
h |
“number of implicit hs” |
>0 |
Y |
|
H |
“total number of Hs” |
1 |
||
r |
“size of smallest SSSR ring” |
>0 |
Y |
|
R |
“number of SSSR rings” |
>0 |
Y |
|
v |
“total valence” |
1 |
Y |
|
x |
“number of ring bonds” |
>0 |
Y |
|
X |
“total degree” |
1 |
Y |
|
z |
“number of heteroatom neighbors” |
>0 |
Y |
extension |
Z |
“number of aliphatic heteroatom neighbors” |
>0 |
Y |
extension |
* |
“any atom” |
|||
+ |
“positive charge” |
1 |
Y |
|
++ |
“+2 charge” |
|||
- |
“negative charge” |
1 |
Y |
|
-- |
“-2 charge” |
|||
^0 |
“S hybridized” |
n/a |
N |
extension |
^1 |
“SP hybridized” |
n/a |
N |
extension |
^2 |
“SP2 hybridized” |
n/a |
N |
extension |
^3 |
“SP3 hybridized” |
n/a |
N |
extension |
^4 |
“SP3D hybridized” |
n/a |
N |
extension |
^5 |
“SP3D2 hybridized” |
n/a |
N |
extension |
Bonds¶
Primitive |
Property |
Notes |
|---|---|---|
“” |
“single or aromatic” |
“unspecified bonds” |
- |
single |
|
= |
double |
|
# |
triple |
|
: |
aromatic |
|
~ |
“any bond” |
|
@ |
“ring bond” |
|
/ |
“directional” |
|
\ |
“directional” |
|
-> |
“dative right” |
extension |
<- |
“dative left” |
extension |
Облигации ¶
Примитивный |
Свойство |
Примечания |
|---|---|---|
[H] |
[#1] |
|
[H+] |
[#1+] |
|
[H,Cl] |
[*H1,Cl] |
|
[HH] |
[*H1;*H1] |
Hs в SMARTS ¶
H в SMARTS интерпретируются как атомы водорода, если эквивалентное выражение атома также будет допустимым SMILES; в противном случае они интерпретируются как запрос на любой атом с одним присоединенным водородом.
Вот несколько примеров:
СМАРТС |
Интерпретация |
|---|---|
[H] |
[#1] |
[H+] |
[#1+] |
[H,Cl] |
[*H1,Cl] |
[HH] |
[*H1;*H1] |
Это несколько сбивает с толку, но соответствует документации Daylight ( https://www.daylight.com/dayhtml/doc/theory/theory.smirks.html ):
Следовательно, одно изменение в интерпретации SMARTS для выражений вида: [<weight>H<charge><map>]. В SMARTS эти выражения теперь интерпретируются как атом водорода, а не как любой атом с одним присоединенным водородом. Все остальные водородные выражения SMARTS сохраняют свои значения до 4.51.
Всегда можно увидеть интерпретацию SMARTS в RDKit, используя DescribeQuery()функцию:
>>> print(Chem.AtomFromSmarts('[H,Cl]').DescribeQuery())
AtomOr
AtomHCount 1 = val
AtomType 17 = val
>>> print(Chem.AtomFromSmarts('[2H+]').DescribeQuery())
AtomAnd
AtomAnd
AtomAtomicNum 1 = val
AtomIsotope 2 = val
AtomFormalCharge 1 = val
Самый безопасный (и понятный) способ включить атомы H в ваши запросы — использовать примитив атомного номера [#1] вместо [H] .
Поддержка и расширения Mol/SDF ¶
RDKit охватывает обширный поднабор функций спецификации V2000 и V3000 CTAB. Этот поднабор должен быть лучше документирован.
- Вот поддерживаемые запросы, не являющиеся атомами элементов:
A: любой тяжелый атом
В: любой тяжелый атом, не являющийся углеродом
*: не указано (интерпретируется как любой атом)
L: (V2000): список атомов
AH: (Расширение ChemAxon) любой атом
QH: (расширение ChemAxon) любой неуглеродный атом
X: (Расширение ChemAxon) галоген
XH: (расширение ChemAxon) галоген или водород
- M: (Расширение ChemAxon) металл («содержит щелочные металлы, щелочноземельные металлы, переходные металлы
металлы, актиноиды, лантаноиды, бедные (основные) металлы, Ge, Sb и Po»)
MH: (расширение ChemAxon) металл или водород
- Вот частичный список поддерживаемых функций:
улучшенная стереохимия (только V3000)
Sgroups: Sgroups читаются и пишутся, но интерпретация их содержимого все еще находится в стадии разработки.
Дативные связки в V2000 (тип 9), несмотря на то, что они не являются частью стандарта, мы поддерживаем их, поскольку они часто появляются в реальных данных.
Поиск кольца и SSSR ¶
[Раздел взят из документа «Начало работы»]
Как уже разглагольствовали другие с большей энергией и красноречием, чем я намеревался, определение наименьшего набора наименьших колец молекулы не является уникальным. В некоторых молекулах с высокой симметрией «истинный» SSSR даст результаты, которые не будут привлекательными. Например, SSSR для кубана содержит только 5 колец, хотя их «очевидно» 6. Эту проблему можно решить, реализовав алгоритм малого (вместо наименьшего ) набора наименьших колец, который возвращает симметричные результаты. Это подход, который мы использовали с RDKit.
Поскольку иногда полезно иметь возможность подсчитать, сколько колец SSSR присутствует в молекуле, существует функция GetSSSR, но она возвращает только количество SSSR, а не потенциально неуникальный набор колец.
Для ситуаций, когда вам просто важно знать, находятся ли атомы/связи в кольцах, RDKit предоставляет функцию
rdkit.Chem.rdmolops.FastFindRings(). Она выполняет обход в глубину графа молекулы и определяет атомы и связи, которые находятся в кольцах.
Стереохимия ¶
Типы поддерживаемой стереохимии ¶
В настоящее время RDKit полностью поддерживает тетраэдрическую атомную стереохимию и цис/транс-стереохимию двойных связей. Имеется частичная поддержка нететраэдрической стереохимии, см. раздел Поддержка нететраэдрической атомной стереохимии .
Идентификация потенциальных стереоатомов/стереосвязей ¶
Начиная с версии 2020.09 в RDKit реализовано два различных способа определения потенциальных стереоатомов/стереосвязей:
Устаревший подход:
AssignStereochemistry(). Этот подход делает разумную работу по распознаванию потенциальных стереоцентров, включая некоторую парастереохимию. Он также имеет побочный эффект назначения приблизительных меток CIP атомам/связям (см. ниже). В настоящее время это алгоритм по умолчанию.Новый подход:
FindPotentialStereo(). Новый подход и точнее (особенно для парастереохимии), и быстрее. Он станет подходом по умолчанию в будущей версии RDKit.
Конкретный пример повышения точности благодаря новому алгоритму:
|
|
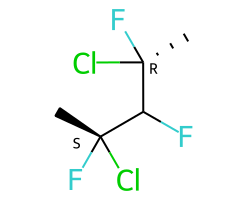
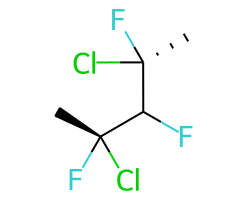
Оба алгоритма распознают, что центральный углерод является потенциальным стереоцентром в молекуле слева, но старый алгоритм не может распознать его как потенциальный стереоцентр в молекуле справа.
Назначение абсолютной стереохимии ¶
Начиная с версии 2020.09 в RDKit реализовано два различных способа назначения абсолютных стереохимических меток (меток CIP):
Устаревший подход использует адаптацию приблизительного алгоритма для назначения кодов CIP, опубликованного Полом Лабутом, [ 12 ] . Алгоритм надежен для определения того, является ли конкретный указанный стереоатом/стереосвязь на самом деле стереоатомом/стереосвязью, но коды CIP, которые он назначает, действительно верны только для простых примеров. Начиная с версии 2020.09 это алгоритм по умолчанию, но он будет изменен в будущей версии RDKit.
Новый подход использует реализацию гораздо более точного алгоритма, [ 13 ] . Новый алгоритм более затратен в вычислительном отношении, чем старый, и не обеспечивает ранжирования атомов CIP (концепция глобального ранжирования атомов не очень хорошо определена в контексте настоящего алгоритма CIP). Если вы заинтересованы в ранжировании всех атомов, чувствительном к хиральности, вы можете использовать канонический код ранжирования атомов.
Стереогенные атомы/связи ¶
Определения потенциальных стереогенных атомов или связей основаны на определениях InChI.
Стереогенные связи ¶
Двойная связь потенциально стереогенна, если у обоих атомов есть по крайней мере два тяжелых атома-соседа и она не присутствует в кольце, содержащем менее восьми атомов.
Например, обе эти двойные связи являются кандидатами на стереохимию:
|
|
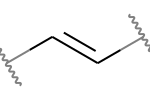
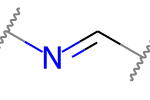
Но это не так:
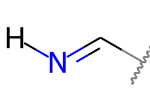
Тетраэдрические стереогенные атомы ¶
Следующие типы атомов являются потенциальными тетраэдрическими стереогенными атомами:
атомы со степенью 4
атомы со степенью 3 и одним неявным H
P или As со степенью 3 или 4
Гибридизированный N SP3 со степенью 3, который не участвует ни в каких сопряженных связях и находится в кольце размером 3 или который является общим для по крайней мере 3 колец (это последнее условие является расширением правил InChI).
S или Se со степенью 3 и общей валентностью 4 или общей валентностью 3 и суммарным зарядом +1.
Краткое описание findPotentialStereo()алгоритма ¶
Определите все потенциальные стереогенные атомы и связи в молекуле. Если их нет, то нам больше ничего делать не нужно.
Для каждого потенциального стереогенного атома: сохранить исходный хиральный тег, а затем установить хиральный тег на CW. Назначить символ атома, который делает этот атом уникальным среди всех остальных (он будет использоваться ниже в алгоритме канонизации)
Для каждой потенциальной стереогенной связи: назначить символ связи, который сделает эту связь уникальной среди всех остальных (он будет использоваться ниже в алгоритме канонизации)
Определить канонический рейтинг атомов, принимая во внимание хиральность, но не разрывая связей. Это использует тот же алгоритм канонизации, который используется для генерации SMILES. [ 14 ]
Удалите хиральный тег из любого потенциального стереогенного атома, имеющего двух соседей с одинаковым рангом, и установите его символ по умолчанию для этого атома.
Установите символ любой двойной связи, которая имеет два атома одинакового ранга, присоединенных к любому концу [ 15 ] , как символ по умолчанию для этой связи
Если шаги 5 и 6 изменили какие-либо атомы или связи, вернитесь к шагу 4.
Добавьте к результатам любой потенциальный стереогенный атом, у которого нет соседей с одинаковым рангом.
Добавьте к результатам любой потенциальный стереогенный атом, который не имеет двух атомов одинакового ранга, прикрепленных к любому из концов [ 15 ].
Верните результаты
Источники информации о стереохимии ¶
Из УЛЫБОК ¶
Атомную стереохимию можно задать с помощью @, @@, @SPи т.д. Потенциальные стереоцентры, для которых не предоставлена информация, — это
ChiralType::CHI_UNSPECIFIED.
Стереохимия двойной связи определяется с помощью /и \для указания направленности соседних одинарных связей. Двойные связи без указанной стереоинформации — это BondStereo::STEREONONE.
Из Мол ¶
Атомная стереохимия может быть задана с помощью клиновидных связей, если присутствуют 2D-координаты. Если присутствуют 3D-координаты, они используются для установки стереохимии для стереогенных атомов. Волнистые связи ( CFG=2в блоках V3000 моль) устанавливают хиральный тег стереогенного начального атома на ChiralType::CHI_UNSPECIFIED.
Стереохимия двойной связи автоматически устанавливается с использованием атомных координат; это справедливо как для 2D, так и для 3D координат. Если стереогенная двойная связь пересекается ( CFG=2в блоках V3000 моль) или имеет смежную волнистую одинарную связь ( CFG=2в блоках V3000 моль), то это будет BondStereo::STEREOANY.
Из CXSMILES ¶
Начальное задание по стереохимии выполняется в соответствии с правилами SMILES (см. выше).
Спецификация w:(волнистая связь) установит стереохимию начального атома на ChiralType::CHI_UNSPECIFIEDи двойных связей на
BondStereo::STEREOANY. Стереохимию кольцевых связей можно задать с помощью t,
c, или ctu.
Если в CXSMILES присутствуют 2D-координаты, атомное стерео можно задать с помощью
`wU`или `wD`для создания клиновидных связей.
Если в CXSMILES присутствуют 3D-координаты, они используются для задания стереохимии для стереогенных атомов и связей. Это заменяет другие спецификации в CXSMILES, за исключением ctuи w.
Поддержка нететраэдрической атомной стереохимии ¶
Начиная с версии 2022.09, RDKit имеет частичную, но развивающуюся поддержку нететраэдрической стереохимии. Статус этой работы отслеживается в этой проблеме github: https://github.com/rdkit/rdkit/issues/4851
Этот код выпускается в предварительном состоянии, чтобы как можно скорее получить обратную связь и начать накапливать опыт работы с этими системами.
Статус по состоянию на релиз 2022.09.1 ¶
"Полный" ¶
(Обратите внимание, что поскольку это новая территория, термин «полный» следует воспринимать с долей скептицизма.)
Основное представление
Анализ SMILES и SMARTS
Генерация 2D-координат
Назначение нететраэдрического стерео из 3D структур
Частичный ¶
Написание SMILES. Сгенерированные SMILES должны быть правильными, но они не являются каноническими.
Генерация 3D-координат. Основы здесь работают, но «хиральность» структур TBP и OH неверна.
Пишем файлы mol. Нужны клиновые связи, чтобы это было сделано
Полностью отсутствует ¶
Расклинивающие связи
Письменные SMARTS
Интеграция поиска подструктуры
Назначение CIP
Канонизация
Очистка стереохимии: распознавание неверных спецификаций стереохимии
Назначение нететраэдрического стерео из 2D структур
Обозначение SMILES ¶
Это обсуждение нотации SMILES во многом основано на документации OpenSMILES: http://opensmiles.org/opensmiles.html . Большое спасибо команде, которая подготовила этот документ, и Джону Мэйфилду за его превосходный инструмент CDK Depict, который я использовал для повторной проверки своей работы.
Представление имеет тег, указывающий, что такое стерео, например @SP, и номер перестановки.
Квадратный плоский ¶
@SP1 |
@SP2 |
@SP3 |
|---|---|---|
|
|
|
У |
4 |
З |
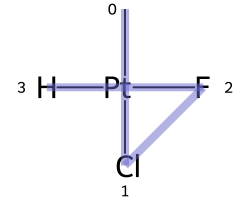
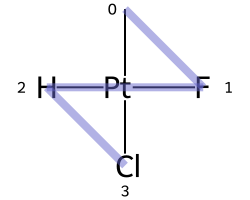
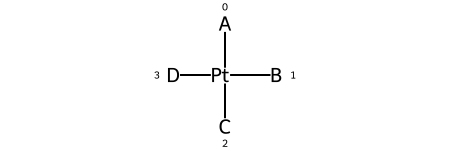
Ниже приведена нумерация лигандов для трех возможных перестановок молекулы образца:
Этикетка |
А |
Б |
С |
Д |
УЛЫБКИ |
|---|---|---|---|---|---|
@SP1 |
0 |
1 |
2 |
3 |
|
@SP2 |
0 |
2 |
1 |
3 |
|
@SP3 |
0 |
1 |
3 |
2 |
|
Тригонально-бипирамидальный ¶
Вот конкретный пример (из документации OpenSMILES):
Ниже приведены метки лигандов и нумерация лигандов для @TB1:
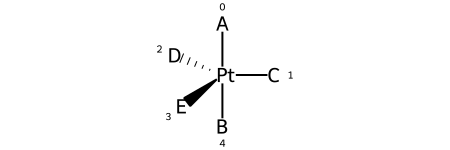
А затем нумерация лигандов для 20 возможных перестановок молекулы образца:
Этикетка |
А |
Б |
С |
Д |
Э |
УЛЫБКИ |
|---|---|---|---|---|---|---|
@TB1 |
0 |
4 |
1 |
2 |
3 |
|
@TB2 |
0 |
4 |
1 |
3 |
2 |
|
@TB3 |
0 |
3 |
1 |
2 |
4 |
|
@TB4 |
0 |
3 |
1 |
4 |
2 |
|
@TB5 |
0 |
2 |
1 |
3 |
4 |
|
@TB6 |
0 |
2 |
1 |
4 |
3 |
|
@TB7 |
0 |
1 |
2 |
3 |
4 |
|
@TB8 |
0 |
1 |
2 |
4 |
3 |
|
@TB9 |
1 |
4 |
0 |
2 |
3 |
|
@TB11 |
1 |
4 |
0 |
3 |
2 |
|
@TB10 |
1 |
3 |
0 |
2 |
4 |
|
@TB12 |
1 |
3 |
0 |
4 |
2 |
|
@TB13 |
1 |
2 |
0 |
3 |
4 |
|
@TB14 |
1 |
2 |
0 |
4 |
3 |
|
@TB15 |
2 |
4 |
0 |
1 |
3 |
|
@TB20 |
2 |
4 |
0 |
3 |
1 |
|
@TB16 |
2 |
3 |
0 |
1 |
4 |
|
@TB19 |
2 |
3 |
0 |
4 |
1 |
|
@TB17 |
3 |
4 |
0 |
1 |
2 |
|
@TB18 |
3 |
4 |
0 |
2 |
1 |
|
Октаэдрический ¶
Вот конкретный пример (изобретенная молекула):
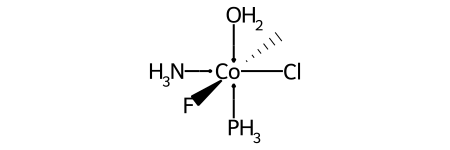
Ниже приведены метки лигандов и нумерация лигандов для @OH1:
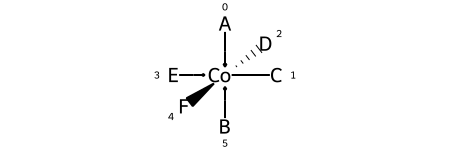
А затем квадратная плоская форма и нумерация лигандов для 30 возможных перестановок молекулы образца:
Этикетка |
СП |
А |
Б |
С |
Д |
Э |
Ф |
УЛЫБКИ |
|---|---|---|---|---|---|---|---|---|
@OH1 |
У |
0 |
5 |
1 |
2 |
3 |
4 |
|
@OH2 |
У |
0 |
5 |
1 |
4 |
3 |
2 |
|
@OH3 |
У |
0 |
4 |
1 |
2 |
3 |
5 |
|
@OH16 |
У |
0 |
4 |
1 |
5 |
3 |
2 |
|
@OH6 |
У |
0 |
3 |
1 |
2 |
4 |
5 |
|
@OH18 |
У |
0 |
3 |
1 |
5 |
4 |
2 |
|
@OH19 |
У |
0 |
2 |
1 |
3 |
4 |
5 |
|
@OH24 |
У |
0 |
2 |
1 |
5 |
4 |
3 |
|
@OH25 |
У |
0 |
1 |
2 |
3 |
4 |
5 |
|
@OH30 |
У |
0 |
1 |
2 |
5 |
4 |
3 |
|
@OH4 |
З |
0 |
5 |
1 |
2 |
4 |
3 |
|
@OH14 |
З |
0 |
5 |
1 |
3 |
4 |
2 |
|
@OH5 |
З |
0 |
4 |
1 |
2 |
5 |
3 |
|
@OH15 |
З |
0 |
4 |
1 |
3 |
5 |
2 |
|
@OH7 |
З |
0 |
3 |
1 |
2 |
5 |
4 |
|
@OH17 |
З |
0 |
3 |
1 |
4 |
5 |
2 |
|
@OH20 |
З |
0 |
2 |
1 |
3 |
5 |
4 |
|
@OH23 |
З |
0 |
2 |
1 |
4 |
5 |
3 |
|
@OH26 |
З |
0 |
1 |
2 |
3 |
5 |
4 |
|
@OH29 |
З |
0 |
1 |
2 |
4 |
5 |
3 |
|
@OH10 |
4 |
0 |
5 |
1 |
4 |
2 |
3 |
|
@OH8 |
4 |
0 |
5 |
1 |
3 |
2 |
4 |
|
@OH11 |
4 |
0 |
4 |
1 |
5 |
2 |
3 |
|
@OH9 |
4 |
0 |
4 |
1 |
3 |
2 |
5 |
|
@OH13 |
4 |
0 |
3 |
1 |
4 |
2 |
4 |
|
@OH12 |
4 |
0 |
3 |
1 |
4 |
2 |
5 |
|
@OH22 |
4 |
0 |
2 |
1 |
5 |
3 |
4 |
|
@OH21 |
4 |
0 |
2 |
1 |
4 |
3 |
5 |
|
@OH28 |
4 |
0 |
1 |
2 |
5 |
3 |
4 |
|
@OH27 |
4 |
0 |
1 |
2 |
4 |
3 |
5 |
|
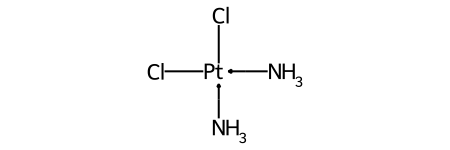
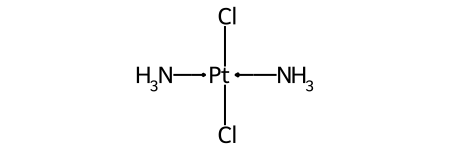
Дублирующие лиганды ¶
Одним из основных различий между нететраэдрической стереохимией и тетраэдрическим вариантом является возможность существования нететраэдрической стереохимии с центральными атомами, имеющими дублирующиеся лиганды.
Классический пример — цис-платин - Cl[Pt@SP1](Cl)(<-[NH3])<-[NH3]- против транс-платина - Cl[Pt@SP2](Cl)(<-[NH3])<-[NH3]-
|
|
|---|---|
|
|
Обработка неявных Hs ¶
Неявные H рассматриваются так же, как в тетраэдрическом стерео: как будто они являются первыми соседями после центрального атома. Таким образом, две улыбки C[Pt@SP1H](Cl)F
и C[Pt@SP1]([H])(Cl)Fсоответствуют одной и той же структуре.
Это также работает с несколькими неявными H: C[Pt@SP1H2]Clи C[Pt@SP1]([H])([H])Clони эквивалентны.
Отсутствующие лиганды ¶
Координационные среды с отсутствующими лигандами рассматриваются так, как если бы отсутствующие лиганды находились в конце упорядочения лигандов. Например, этот изобретенный комплекс может быть представлен с помощью SMILES O[Mn@OH1](Cl)(C)(N)F.
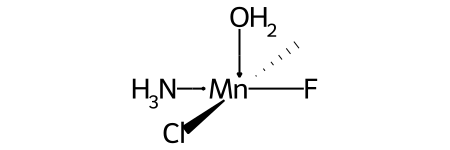
Сравните это с УЛЫБКАМИ для соответствующего комплекса, показанного выше при обсуждении @OHстерео.
Обработка химических реакций ¶
Реакция SMARTS ¶
Не УМЫЛКИ [ 1 ] , не УМЫЛКИ-РЕАКЦИИ [ 2 ] , а производные от УМНОСТИ [ 3 ] .
Общая грамматика для реакции SMARTS такова:
реакция ::= реагенты ">>" продукты
реагенты ::= молекулы
продукты ::= молекулы
молекулы ::= молекула
молекулы "." молекула
молекула ::= допустимая строка SMARTS без символов "."
"(" допустимая строка SMARTS без символов "." ")"
Некоторые особенности ¶
Отображенные фиктивные атомы в шаблоне продукта заменяются соответствующим атомом в реагенте:
>>> from rdkit.Chem import AllChem
>>> rxn = AllChem.ReactionFromSmarts('[C:1]=[O,N:2]>>[C:1][*:2]')
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('CC=O'),))[0]]
['CCO']
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('CC=N'),))[0]]
['CCN']
но неотображенные фиктивные атомы остаются фиктивными:
>>> rxn = AllChem.ReactionFromSmarts('[C:1]=[O,N:2]>>*[C:1][*:2]')
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('CC=O'),))[0]]
['*C(C)O']
«Любые» связи в продуктах заменяются соответствующей связью в реагенте:
>>> rxn = AllChem.ReactionFromSmarts('[C:1]~[O,N:2]>>*[C:1]~[*:2]')
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('C=O'),))[0]]
['*C=O']
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('CO'),))[0]]
['*CO']
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('C#N'),))[0]]
['*C#N']
Внутримолекулярные реакции можно выразить гибко, включив реагенты в скобки. Это показано в этом примере метатезиса с замыканием кольца [ 5 ] :
>>> rxn = AllChem.ReactionFromSmarts("([C:1]=[C;H2].[C:2]=[C;H2])>>[*:1]=[*:2]")
>>> m1 = Chem.MolFromSmiles('C=CCOCC=C')
>>> ps = rxn.RunReactants((m1,))
>>> Chem.MolToSmiles(ps[0][0])
'C1=CCOC1'
Хиральность ¶
В этом разделе описывается, как обрабатывается информация о хиральности в определении реакции. Последовательный пример, этерификация вторичных спиртов, используется на протяжении всего [ 6 ] .
Если в определении реакции отсутствует хиральная информация, стереохимия реагентов сохраняется, как и членство в расширенных стереогруппах:
>>> alcohol1 = Chem.MolFromSmiles('CC(CCN)O')
>>> alcohol2 = Chem.MolFromSmiles('C[C@H](CCN)O')
>>> alcohol3 = Chem.MolFromSmiles('C[C@@H](CCN)O')
>>> acid = Chem.MolFromSmiles('CC(=O)O')
>>> rxn = AllChem.ReactionFromSmarts('[CH1:1][OH:2].[OH][C:3]=[O:4]>>[C:1][O:2][C:3]=[O:4]')
>>> ps=rxn.RunReactants((alcohol1,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)OC(C)CCN'
>>> ps=rxn.RunReactants((alcohol2,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol3,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@@H](C)CCN'
Вы получите тот же результат (сохранение стереохимии), если отображенный атом имеет одинаковую хиральность как в реагентах, так и в продуктах:
>>> rxn = AllChem.ReactionFromSmarts('[C@H1:1][OH:2].[OH][C:3]=[O:4]>>[C@:1][O:2][C:3]=[O:4]')
>>> ps=rxn.RunReactants((alcohol1,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)OC(C)CCN'
>>> ps=rxn.RunReactants((alcohol2,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol3,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@@H](C)CCN'
Картографированный атом с различной хиральностью в реагентах и продуктах приводит к инверсии стереохимии:
>>> rxn = AllChem.ReactionFromSmarts('[C@H1:1][OH:2].[OH][C:3]=[O:4]>>[C@@:1][O:2][C:3]=[O:4]')
>>> ps=rxn.RunReactants((alcohol1,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)OC(C)CCN'
>>> ps=rxn.RunReactants((alcohol2,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol3,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
Если отображенный атом имеет хиральность, указанную в реагентах, но не в продуктах, реакция разрушает хиральность в этом центре:
>>> rxn = AllChem.ReactionFromSmarts('[C@H1:1][OH:2].[OH][C:3]=[O:4]>>[C:1][O:2][C:3]=[O:4]')
>>> ps=rxn.RunReactants((alcohol1,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)OC(C)CCN'
>>> ps=rxn.RunReactants((alcohol2,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)OC(C)CCN'
>>> ps=rxn.RunReactants((alcohol3,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)OC(C)CCN'
И, наконец, если хиральность указана в продуктах, но не в реагентах, то в реакции создается стереоцентр с указанной хиральностью:
>>> rxn = AllChem.ReactionFromSmarts('[CH1:1][OH:2].[OH][C:3]=[O:4]>>[C@:1][O:2][C:3]=[O:4]')
>>> ps=rxn.RunReactants((alcohol1,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol2,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol3,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
Это не имеет смысла, если в определение реакции не включить немного больше контекста вокруг стереоцентра:
>>> rxn = AllChem.ReactionFromSmarts('[CH3:5][CH1:1]([C:6])[OH:2].[OH][C:3]=[O:4]>>[C:5][C@:1]([C:6])[O:2][C:3]=[O:4]')
>>> ps=rxn.RunReactants((alcohol1,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol2,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol3,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
Обратите внимание, что спецификация хиральности не используется как часть запроса: молекула без указанной хиральности может соответствовать реагенту с указанной хиральностью.
В общем, реакционная машина пытается сохранить как можно больше стереохимической информации. Это работает, когда образуется одна новая связь с хиральным центром:
>>> rxn = AllChem.ReactionFromSmarts('[C:1][C:2]-O>>[C:1][C:2]-S')
>>> alcohol2 = Chem.MolFromSmiles('C[C@@H](O)CCN')
>>> ps=rxn.RunReactants((alcohol2,))
>>> Chem.MolToSmiles(ps[0][0],True)
'C[C@@H](S)CCN'
Однако этот метод не работает, если образуются две или более связей:
>>> rxn = AllChem.ReactionFromSmarts('[C:1][C:2](-O)-F>>[C:1][C:2](-S)-Cl')
>>> alcohol = Chem.MolFromSmiles('C[C@@H](O)F')
>>> ps=rxn.RunReactants((alcohol,))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(S)Cl'
В этом случае просто недостаточно информации, чтобы сохранить информацию. Вы можете помочь, предоставив картографическую информацию:
Некоторые предостережения Мы сделали этот код настолько надежным, насколько это возможно, но это нетривиальная проблема, и, безусловно, можно получить неожиданные результаты.
Все становится сложнее, если порядок атомов вокруг хирального центра изменяется в реакции SMARTS. Вот некоторые из ситуаций, которые в настоящее время обрабатываются правильно.
Переупорядочение соседей, но число и атомные отображения соседей остаются постоянными. В этом случае нет инверсии хиральности, даже если хиральный тег на хиральном атоме меняется между реагентами и продуктами:
>>> rxn = AllChem.ReactionFromSmarts('[C:1][C@:2]([F:3])[Br:4]>>[C:1][C@@:2]([S:4])[F:3]')
>>> mol = Chem.MolFromSmiles('C[C@@H](F)Br')
>>> ps=rxn.RunReactants((mol,))
>>> Chem.MolToSmiles(ps[0][0],True)
'C[C@@H](F)S'
Добавление соседа к хиральному атому.
>>> rxn = AllChem.ReactionFromSmarts('[C:1][C@H:2]([F:3])[Br:4]>>[C:1][C@@:2](O)([F:3])[Br:4]')
>>> mol = Chem.MolFromSmiles('C[C@@H](F)Br')
>>> ps=rxn.RunReactants((mol,))
>>> Chem.MolToSmiles(ps[0][0],True)
'C[C@](O)(F)Br'
Удаление соседа из хирального атома.
>>> rxn = AllChem.ReactionFromSmarts('[C:1][C@:2](O)([F:3])[Br:4]>>[C:1][C@@H:2]([F:3])[Br:4]')
>>> mol = Chem.MolFromSmiles('C[C@@](O)(F)Br')
>>> ps=rxn.RunReactants((mol,))
>>> Chem.MolToSmiles(ps[0][0],True)
'C[C@H](F)Br'
Правила и предупреждения ¶
Включайте информацию карты атома в конец запроса атома. Так же как [C,N,O:1] или [C;R:1].
Не забывайте, что неопределенные связи в SMARTS являются либо одинарными, либо ароматическими. Порядки связей в шаблонах продуктов назначаются при построении самого шаблона продукта, и не всегда можно сказать, должна ли связь быть одинарной или ароматической:
>>> rxn = AllChem.ReactionFromSmarts('[#6:1][#7,#8:2]>>[#6:1][#6:2]')
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('C1NCCCC1'),))[0]]
['C1CCCCC1']
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('c1ncccc1'),))[0]]
['c1ccccc-1']
So if you want to copy the bond order from the reactant, use an “Any” bond:
>>> rxn = AllChem.ReactionFromSmarts('[#6:1][#7,#8:2]>>[#6:1]~[#6:2]')
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('c1ncccc1'),))[0]]
['c1ccccc1']
Формат файла определения функций ¶
Файл FDef содержит всю информацию, необходимую для определения набора химических характеристик. Он содержит определения типов характеристик, которые определяются из запросов, созданных с использованием языка Daylight SMARTS. [ 3 ] Файл FDef может также опционально включать определения типов атомов, которые используются для того, чтобы сделать определения характеристик более читаемыми.
Химические характеристики ¶
Химические характеристики определяются типом характеристики и семейством характеристик. Семейство характеристик представляет собой общую классификацию характеристики (например, «Донор водородной связи» или «Ароматический»), в то время как тип характеристики предоставляет дополнительную информацию с более высоким разрешением о характеристиках. Сопоставление фармакофоров выполняется с использованием семейств характеристик. Каждый тип характеристики содержит следующую информацию:
Шаблон SMARTS, описывающий атомы (один или несколько), соответствующие типу объекта.
Веса, используемые для определения положения объекта на основе положений его определяющих атомов.
Синтаксис файла FDef ¶
Определения AtomType ¶
Определение AtomType позволяет назначить сокращенное имя, которое будет использоваться вместо строки SMARTS, определяющей запрос атома. Это позволяет сделать файлы FDef гораздо более читаемыми. Например, определение неполярного атома углерода следующим образом:
AtomType Carbon_NonPolar [C&!$(C=[O,N,P,S])&!$(C#N)]
создает новое имя, которое может быть использовано в любом другом месте файла FDef, где было бы полезно использовать этот SMARTS. Чтобы сослаться на AtomType, просто включите его имя в фигурные скобки. Например, этот отрывок из файла FDef определяет другой тип атома - Hphobe - который ссылается на определение Carbon_NonPolar:
AtomType Carbon_NonPolar [C&!$(C=[O,N,P,S])&!$(C#N)]
AtomType Hphobe [{Carbon_NonPolar},c,s,S&H0&v2,F,Cl,Br,I]
Обратите внимание, что {Carbon_NonPolar}в новом определении AtomType используется без каких-либо дополнительных украшений (квадратные скобки или рекурсивные маркеры SMARTS не требуются).
Повторение AtomType приводит к объединению двух определений с помощью оператора SMARTS “,” (или). Вот пример:
AtomType d1 [N&!H0]
AtomType d1 [O&!H0]
Это эквивалентно:
AtomType d1 [N&!H0,O&!H0]
Что эквивалентно более эффективному:
AtomType d1 [N,O;!H0]
Обратите внимание , что эти примеры, как правило, используют оператор SMARTS с высоким приоритетом и «&», а не с низким приоритетом и «;». Это может быть важно, когда AtomTypes объединяются или повторяются. Оператор SMARTS «,» имеет более высокий приоритет, чем «;», поэтому определения, использующие «;», могут привести к неожиданным результатам.
Также возможно определение отрицательных запросов AtomType:
AtomType d1 [N,O,S]
AtomType !d1 [H0]
Отрицательный запрос объединяется с первым, чтобы создать определение, идентичное этому:
AtomType d1 [!H0;N,O,S]
Обратите внимание, что в начало запроса добавляется отрицательный AtomType.
Определения функций ¶
Определение функции сложнее определения AtomType и занимает несколько строк:
DefineFeature HDonor1 [N,O;!H0]
Family HBondDonor
Weights 1.0
EndFeature
Первая строка определения функции включает тип функции и строку SMARTS, определяющую функцию. Следующие две строки (порядок не важен) определяют семейство функции и ее атомные веса (разделенный запятыми список той же длины, что и количество атомов, определяющих функцию). Атомные веса используются для расчета местоположений функции на основе взвешенного среднего положения атома, определяющего функцию. Более подробная информация об этом приведена ниже. Последняя строка определения функции должна быть EndFeature. Совершенно допустимо смешивать определения AtomType с определениями функций в файле FDef. Единственное правило заключается в том, что AtomTypes должны быть определены до того, как на них будут ссылаться.
Дополнительные примечания по синтаксису: ¶
Любая строка, начинающаяся с символа #, считается комментарием и будет игнорироваться.
Символ обратной косой черты, , в конце строки является символом продолжения, он указывает, что данные из этой строки продолжаются на следующей строке файла. Пробел в начале этих дополнительных строк игнорируется. Например, это определение AtomType:
AtomType tButylAtom [$([C;!R](-[CH3])(-[CH3])(-[CH3])),\ $([CH3](-[C;!R](-[CH3])(-[CH3])))]
точно эквивалентно этому:
AtomType tButylAtom [$([C;!R](-[CH3])(-[CH3])(-[CH3])),$([CH3](-[C;!R](-[CH3])(-[CH3])))]
(хотя первую форму гораздо легче читать!)
Веса атомов и расположение элементов ¶
Часто задаваемые вопросы ¶
Что произойдет, если тип объекта будет повторяться в файле? Вот пример:
DefineFeature HDonor1 [O&!H0] Family HBondDonor Weights 1.0 EndFeature DefineFeature HDonor1 [N&!H0] Family HBondDonor Weights 1.0 EndFeature
В этом случае оба определения типа функции HDonor1 будут активны. Это функционально идентично:
DefineFeature HDonor1 [O,N;!H0] Family HBondDonor Weights 1.0 EndFeature
Однако формулировка этого определения признака с дублирующим типом признака значительно менее эффективна и более запутанна, чем более простое комбинированное определение.
Представление отпечатков ДÅктიЛიскооРи фармакофоров ¶
В схеме RDKit битовые идентификаторы в отпечатках ДÅктიЛიскооРи фармакофоров не хэшируются: каждый бит соответствует определенной комбинации признаков и расстояний. Заданный битовый идентификатор может быть преобразован обратно в соответствующие типы признаков и расстояния для обеспечения интерпретации. Иллюстрация для 2D-фармакофоров показана на рисунке 1: Нумерация битов в отпечатках ДÅктიЛიскооРи фармакофоров .

Рисунок 1: Нумерация битов в отпечатках ДÅктიЛიскооРи фармакофоров ¶
Сопоставление атомов в запросах подструктур ¶
При выполнении сопоставлений подструктур для запросов, полученных из SMARTS, правила, определяющие, какие атомы в молекуле должны соответствовать каким атомам в запросе, четко определены. [#smarts]_ То же самое не обязательно происходит, когда молекула запроса получена из блока mol или SMILES.
Общее правило, используемое в RDKit, заключается в том, что если вы не указываете свойство в запросе, то оно не используется как часть критериев соответствия и H игнорируются. Это приводит к следующему поведению:
Молекула |
Запрос |
Соответствовать |
|---|---|---|
CCO |
CCO |
Да |
CC[O-] |
CCO |
Да |
CCO |
CC[O-] |
Нет |
CC[O-] |
CC[O-] |
Да |
CC[O-] |
СС[ОН] |
Да |
CCOC |
СС[ОН] |
Да |
CCOC |
CCO |
Да |
КСС |
КСС |
Да |
СС[14С] |
КСС |
Да |
КСС |
СС[14С] |
Нет |
СС[14С] |
СС[14С] |
Да |
ОКО |
С |
Да |
ОКО |
[Ч] |
Нет |
ОКО |
[СН2] |
Нет |
ОКО |
[СН3] |
Нет |
ОКО |
О[СН3] |
Да |
О[СН2]О |
С |
Да |
О[СН2]О |
[СН2] |
Нет |
Демонстрируется здесь:
>>> Chem.MolFromSmiles('CCO').HasSubstructMatch(Chem.MolFromSmiles('CCO'))
True
>>> Chem.MolFromSmiles('CC[O-]').HasSubstructMatch(Chem.MolFromSmiles('CCO'))
True
>>> Chem.MolFromSmiles('CCO').HasSubstructMatch(Chem.MolFromSmiles('CC[O-]'))
False
>>> Chem.MolFromSmiles('CC[O-]').HasSubstructMatch(Chem.MolFromSmiles('CC[O-]'))
True
>>> Chem.MolFromSmiles('CC[O-]').HasSubstructMatch(Chem.MolFromSmiles('CC[OH]'))
True
>>> Chem.MolFromSmiles('CCOC').HasSubstructMatch(Chem.MolFromSmiles('CC[OH]'))
True
>>> Chem.MolFromSmiles('CCOC').HasSubstructMatch(Chem.MolFromSmiles('CCO'))
True
>>> Chem.MolFromSmiles('CCC').HasSubstructMatch(Chem.MolFromSmiles('CCC'))
True
>>> Chem.MolFromSmiles('CC[14C]').HasSubstructMatch(Chem.MolFromSmiles('CCC'))
True
>>> Chem.MolFromSmiles('CCC').HasSubstructMatch(Chem.MolFromSmiles('CC[14C]'))
False
>>> Chem.MolFromSmiles('CC[14C]').HasSubstructMatch(Chem.MolFromSmiles('CC[14C]'))
True
>>> Chem.MolFromSmiles('OCO').HasSubstructMatch(Chem.MolFromSmiles('C'))
True
>>> Chem.MolFromSmiles('OCO').HasSubstructMatch(Chem.MolFromSmiles('[CH]'))
False
>>> Chem.MolFromSmiles('OCO').HasSubstructMatch(Chem.MolFromSmiles('[CH2]'))
False
>>> Chem.MolFromSmiles('OCO').HasSubstructMatch(Chem.MolFromSmiles('[CH3]'))
False
>>> Chem.MolFromSmiles('OCO').HasSubstructMatch(Chem.MolFromSmiles('O[CH3]'))
True
>>> Chem.MolFromSmiles('O[CH2]O').HasSubstructMatch(Chem.MolFromSmiles('C'))
True
>>> Chem.MolFromSmiles('O[CH2]O').HasSubstructMatch(Chem.MolFromSmiles('[CH2]'))
False
Общие («Маркуша») запросы в сопоставлении подструктур ¶
Примечание. В этом разделе описываются функциональные возможности, добавленные в версии RDKit 2022.03.1 .
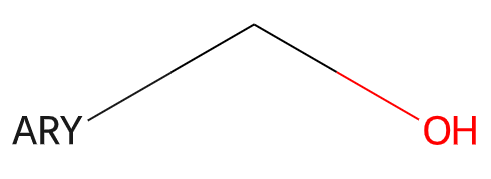
RDKit поддерживает набор общих запросов, используемых как часть систем Beilstein и Reaxys. Вот пример:
Информация о генерических запросах может быть считана из блоков CXSMILES или V3000 Mol (как SUP SGroups) и затем вызвана функция Chem.SetGenericQueriesFromProperties() с молекулой, которую нужно изменить, в качестве аргумента. Эти функции не используются по умолчанию при выполнении запросов подструктур, но могут быть включены, если установить опцию SubstructMatchParameters.useGenericMatchers в значение True
Вот пример использования этих функций:
>>> q = Chem.MolFromSmarts('OC* |$;;ARY$|')
>>> Chem.SetGenericQueriesFromProperties(q)
>>> Chem.MolFromSmiles('C1CCCCC1CO').HasSubstructMatch(q)
True
>>> Chem.MolFromSmiles('c1ccccc1CO').HasSubstructMatch(q)
True
>>> ps = Chem.SubstructMatchParameters()
>>> ps.useGenericMatchers = True
>>> Chem.MolFromSmiles('C1CCCCC1CO').HasSubstructMatch(q,ps)
False
>>> Chem.MolFromSmiles('c1ccccc1CO').HasSubstructMatch(q,ps)
True
Вот поддерживаемые группы и краткое описание того, что они означают:
Алкил (АЛК)
алкильные боковые цепи (не атом H)
АлкилH (ALH)
алкильные боковые цепи, включающие атом H
Алкенил (AEL)
алкенильные боковые цепи
АлкенилH (AEH)
алкенильные боковые цепи или атом H
Алкинил (AYL)
алкинильные боковые цепи
АлкинилH (AYH)
алкинильные боковые цепи или атом H
Алкокси (AOX)
алкокси боковые цепи
АлкоксиH (AOH)
алкокси боковые цепи или атом H
Карбоциклические (CBC)
карбоциклические боковые цепи
КарбоциклическийH (CBH)
карбоциклические боковые цепи или атом H
Карбоциклоалкил (CAL)
циклоалкильные боковые цепи
КарбоциклоалкилH (CAH)
циклоалкильные боковые цепи или атом H
Карбоциклоалкенил (CEL)
циклоалкенильные боковые цепи
КарбоциклоалкенилH (CEH)
циклоалкенильные боковые цепи или атом H
Карбоарил (ARY)
полностью углеродные арильные боковые цепи
КарбоарилH (ARH)
полностью углеродные арильные боковые цепи или атом H
Циклический (CYC)
циклические боковые цепи
ЦиклическийH (CYH)
циклические боковые цепи или атом H
Ациклический (ACY)
ациклические боковые цепи (не атом H)
АциклическийH (ACH)
ациклические боковые цепи или атом H
Карбоациклические (ABC)
ациклические боковые цепи, состоящие из всех атомов углерода
КарбоациклическийH (ABH)
ациклические боковые цепи, состоящие из всех атомов углерода или атома H
Гетероациклические (AHC)
ациклические боковые цепи с по крайней мере одним гетероатомом
ГетероациклическийH (AHH)
ациклические боковые цепи с по крайней мере одним гетероатомом или атомом H
Гетероциклические (CHC)
циклические боковые цепи с по крайней мере одним гетероатомом
ГетероциклическийH (CHH)
циклические боковые цепи с по крайней мере одним гетероатомом или атомом H
Гетероарил (HAR)
арильные боковые цепи с по крайней мере одним гетероатомом
ГетероарилH (HAH)
арильные боковые цепи с по крайней мере одним гетероатомом или атомом H
NoCarbonRing (CXX)
кольцо, не содержащее атомов углерода
NoCarbonRingH (CXH)
кольцо, не содержащее атомов углерода или атома H
Группа (Г)
любая группа (не атом H)
ГруппаH (GH)
любая группа (включая атом H)
Группа* (Г*)
любая группа с замыканием кольца
ГруппаH* (GH*)
любая группа с замыканием кольца или атомом H
Более подробное описание смотрите в документации к файлу C++ GenericGroups.h.
Молекулярная санитарная обработка ¶
Все функции анализа молекул по умолчанию выполняют операцию «санации» считанных молекул. Идея состоит в том, чтобы генерировать полезные вычисляемые свойства (такие как гибридизация, членство в кольце и т. д.) для остальной части кода и гарантировать, что молекулы «разумны»: что они могут быть представлены октетно-полными структурами точек Льюиса.
Вот необходимые шаги в указанном порядке.
clearComputedProps: удаляет все вычисляемые свойства, которые уже существуютна молекуле и ее атомах и связях. Этот шаг выполняется всегда.
cleanUp: стандартизирует небольшое количество нестандартных валентных состояний. Операции очистки:
Нейтральные 5-валентные N с двойными связями с Os преобразуются в цвиттер-ионную форму. Пример:
N(=O)=O -> [N+](=O)O-]Нейтральные 5-валентные N с тройными связями с другим N преобразуются в цвиттер-ионную форму. Пример:
C-N=N#N -> C-N=[N+]=[N-]Нейтральный 5-валентный фосфор с одной двойной связью с O и другой с C или P преобразуется в цвиттер-ионную форму. Пример:
C=P(=O)O -> C=[P+]([O-])OНейтральные Cl, Br или I с исключительно O-соседями и валентностью 3, 5 или 7 преобразуются в цвиттер-ионную форму. Это касается таких веществ, как хлорноватая кислота, хлорноватая кислота и хлорная кислота. Пример:
O=Cl(=O)O -> [O-][Cl+2][O-]OЭтот шаг не должен приводить к возникновению исключений.
cleanUpOrganometallics: стандартизирует небольшое количество нестандартных ситуаций, встречающихся в металлоорганических соединениях. Операции по очистке:
заменяет одинарные связи от гипервалентных атомов к металлам на дативные связи.
Этот шаг не должен приводить к возникновению исключений.
updatePropertyCache: вычисляет явные и неявные валентности для всех атомов. Это создает исключения для атомов в состояниях валентности выше разрешенной. Этот шаг выполняется всегда, но если его «пропустить», тест на нестандартные валентности не будет выполнен.
symmetrizeSSSR: вызывает алгоритм симметризированного наименьшего набора наименьших колец (обсуждается в документе «Начало работы»).
Kekulize: преобразует ароматические кольца в форму Кекуле. Вызовет исключение, если кольцо не может быть кекулировано или если ароматические связи обнаружены вне колец.
assignRadicals: определяет количество радикальных электронов (если таковые имеются) на каждом атоме.
setAromaticity: идентифицирует ароматические кольца и кольцевые системы (см. выше), устанавливает ароматический флаг для атомов и связей, устанавливает порядок связей как ароматический.
setConjugation: определяет, какие связи сопряжены
setHybridization: вычисляет состояние гибридизации каждого атома
cleanupChirality: удаляет хиральные метки из атомов, которые не находятся в состоянии sp3-гибридизации.
adjustHs: добавляет явные Hs, где необходимо сохранить химию. Обычно это требуется для гетероатомов в ароматических кольцах. Классический пример — атом азота в пирроле.
updatePropertyCache: пересчитывает явные и неявные валентности всех атомов. Это создает исключения для атомов в состояниях валентности выше разрешенных. Этот шаг необходим для обнаружения некоторых пограничных случаев, когда входные атомы с нефизическими валентностями принимаются, если они помечены как ароматические.
Отдельные шаги можно включать и выключать при вызове
MolOps::sanitizeMolили Chem.SanitizeMol.
Расчет валентности и допустимые валентности ¶
По умолчанию RDKit довольно строго применяет допустимые валентности при очистке структур (это делается на этапе очистки updatePropertyCache ): атомы, имеющие явную валентность (сумма указанных порядков связей + указанное количество H), превышающую максимально допустимую валентность для элемента, вызовут исключение.
Допустимые валентности элементов (по состоянию на 1 сентября 2024 г.):
Н 1
Он 0
Ли 1 -1
Быть 2
Б 3
С 4
Н 3
О 2
Ф 1
Не 0
На 1,-1
Мг 2,-1
Ал 3
Си 4
П 3,5
С 2,4,6
Кл 1
Ар 0
К 1,-1
Са 2,-1
Га 3
Ge 4
Как 3,5
Сэ 2,4,6
Бр 1
Кр 0
Рб 1,-1
Ср 2,-1
В 3
Сн 2,4
Сб 3,5
Те 2,4,6
Я 1,3,5
Хе 0,2,4,6
Cs 1,-1
Ба 2,-1
Тл-1
Свинец 2,4
Би 3,5
По 2,4,6
В 1,3,5
Рн 0
Элементы, не указанные в таблице, имеют валентность -1 .
Допустимая валентность -1 указывает на то, что элемент может иметь любое значение валентности. Неявные Hs не будут добавлены к атомам с возможной валентностью -1 , когда явная валентность превышает максимальное указанное значение. Так, например, атом Mg с одинарной связью с ним (явная валентность = 1) будет иметь один неявный добавленный H, в то время как атом Mg с тремя связями не будет иметь неявных добавленных Hs. Атомы, у которых единственная разрешенная валентность равна -1, никогда не будут иметь неявных добавленных Hs.
Допустимые валентности заряженных атомов рассчитываются путем рассмотрения допустимых валентностей изоэлектронного элемента. Например, N+ имеет те же допустимые валентности, что и C , в то время как N- имеет те же допустимые валентности, что и O. P -2 , S- , As-2 и Se- являются особыми случаями: все они имеют допустимые валентности 1, 3 и 5.
Поддержка JSON ¶
RDKit поддерживает запись/чтение из двух тесно связанных форматов JSON: commonchem ( https://github.com/CommonChem/CommonChem ) и rdkitjson. commonchem — это хорошо документированный формат, разработанный для эффективного обмена между молекулярными инструментами. rdkitjson — это расширение commonchem, которое включает дополнительные функции, позволяющие сериализовать молекулы RDKit в JSON. Расширения в rdkitjson — улучшенные стерео и группы веществ — в целом полезны, поэтому легко представить, что они будут интегрированы в commonchem в какой-то момент в будущем.
Списки молекул можно преобразовать в JSON с помощью MolInterchange::MolsToJSONData()(C++) или Chem.MolsToJSONData()(Python). Эти вызовы принимают необязательный объект параметров, который можно использовать для указания того, генерируется ли commonchem или rdkitjson. По умолчанию генерируется rdkitjson.
Данные JSON можно преобразовать обратно в молекулы RDKit с помощью MolInterchange::JSONDataToMols()(C++) или Chem.JSONDataToMols()(Python). Парсер автоматически определит, работает ли он с commonchem или rdkitjson.
Формат rdkitjson ¶
Улучшенное стерео ¶
Вот rdkitjson-представление стереогрупп молекулы :C[C@@H]1C([C@H](O)F)O[C@H](C)C([C@@H](O)F)[C@@H]1C |a:7,o1:3,10,&1:1,&2:13|
'stereoGroups': [{'type': 'abs', 'atoms': [7]},
{'type': 'or', 'atoms': [3, 10]},
{'type': 'and', 'atoms': [1]},
{'type': 'and', 'atoms': [13]}],
Группы веществ ¶
Вот представление SUPгруппы веществ в rdkitjson:
'substanceGroups': [{'properties': {'TYPE': 'SUP',
'index': 1,
'LABEL': 'Boc',
'DATAFIELDS': '[]'},
'atoms': [7, 8, 9, 10, 11, 12, 13],
'bonds': [8],
'brackets': [[[6.24, -2.9, 0.0], [6.24, -2.9, 0.0], [0.0, 0.0, 0.0]]],
'cstates': [{'bond': 8, 'vector': [0.0, 0.82, 0.0]}],
'attachPoints': [{'aIdx': 12, 'lvIdx': 5, 'id': '1'}]}],
и один для SRUгруппы:
'substanceGroups': [{'properties': {'TYPE': 'SRU',
'index': 1,
'CONNECT': 'HT',
'LABEL': 'n',
'DATAFIELDS': '[]'},
'atoms': [2, 1, 4],
'bonds': [2, 0],
'brackets': [[[-3.9538, 4.3256, 0.0],
[-3.0298, 2.7252, 0.0],
[0.0, 0.0, 0.0]],
[[-5.4618, 2.8611, 0.0],
[-6.3858, 4.4615, 0.0],
[0.0, 0.0, 0.0]]]}],
Подробности реализации ¶
«Волшебные» значения свойств ¶
Следующие значения свойств регулярно используются в кодовой базе RDKit и могут быть полезны для клиентского кода.
ROMol (Mol в Python) ¶
Имя свойства |
Использовать |
|---|---|
MolFileКомментарии |
Прочитано/записано в строку комментариев CTAB. |
MolFileInfo |
Прочитано/записано в информационную строку CTAB. |
_MolFileChiralFlag |
Считывание/запись в хиральный флаг CTAB. |
_Имя |
Читается/записывается в строку имени CTAB. |
_smilesAtomOutputOrder |
Порядок, в котором атомы были записаны в SMILES |
_smilesBondOutputOrder |
Порядок, в котором облигации были выписаны на SMILES |
Атом ¶
Имя свойства |
Использовать |
|---|---|
_CIPCode |
CIP-код (R или S) атома |
_CIPRank |
целочисленный CIP-ранг атома |
_Возможная хиральность |
установить, является ли атом возможным хиральным центром |
_MolFileRLabel |
Целочисленная метка группы R для атома, считываемая из/записываемая в CTAB. |
_ReactionDegreeИзменено |
устанавливается на атоме в шаблоне продукта реакции, если его степень изменяется в реакции |
_защищенный |
атомы с этим набором свойств не будут рассматриваться как соответствующие запросам реагентов в реакциях |
фиктивныйМеток |
(на фиктивных атомах) считывается/записывается в CTAB как символ атома |
molAtomMapNumber |
номер карты атома для атома, считываемый из/записываемый в SMILES и CTAB |
molfileПсевдоним |
псевдоним файла mol для атома (следует за тегами A), считывается из/записывается в CTAB |
molFileValue |
значение файла mol для атома (следует за тегами V), считываемое из/записываемое в CTAB |
molFileInversionFlag |
используется для обозначения того, изменяется ли стереохимия атома в ходе реакции, считывается из/записывается в CTAB, определяется автоматически из SMILES |
molRxnКомпонент |
к какому компоненту реакции принадлежит атом, считывается из/записывается в CTAB |
molRxnRole |
какую роль играет атом в реакции (1=реагент, 2=продукт, 3=агент), считывается из/записывается в CTAB |
улыбкиСимвол |
определяет символ, который будет записан в SMILES для атома |
Безопасность потока и RDKit ¶
При написании RDKit мы попытались гарантировать, что код будет работать в многопоточной среде, избегая использования глобальных переменных и т. д. Однако сделать код потокобезопасным — не совсем тривиальная задача, поэтому, несомненно, есть некоторые пробелы. В этом разделе описывается, какие части кодовой базы были явно протестированы на потокобезопасность.
- Примечание: за исключением небольшого количества методов/функций
которые принимают
numThreadsаргумент, этот раздел не применяется к использованию RDKit из потоков Python. Boost.Python гарантирует, что только один поток вызывает код C++ в любой момент. Чтобы получить параллельное выполнение в Python, используйте модуль multiprocessing или один из других стандартных подходов Python для этого.
Что было протестировано ¶
Считывание молекул из блоков SMILES/SMARTS/Mol
Запись молекул в блоки SMILES/SMARTS/Mol (см. ниже)
Генерация 2D-координат
Генерация 3D-конформаций с помощью кода дистанционной геометрии
Оптимизация молекул с помощью UFF или MMFF
Создание ДÅктიЛიскооРи
Калькуляторы дескрипторов в $RDBASE/Code/GraphMol/Descriptors
Поиск подструктуры (Примечание: если молекула запроса содержит рекурсивные запросы, ее может быть небезопасно использовать одновременно в нескольких потоках, см. ниже)
Код подграфа
Код ChemTransforms
Код химических реакций
Код Open3DAlign
Код чертежа MolDraw2D
Код InChI с InChI IUPAC v1.06
Известные проблемы ¶
MolSuppliers (например, SDMolSupplier, SmilesMolSupplier?) изменяют свое внутреннее состояние при считывании молекулы. Небезопасно использовать одного поставщика в более чем одном потоке.
Поиск подструктуры с использованием молекул запросов, включающих рекурсивные запросы. Рекурсивные запросы изменяют свое внутреннее состояние при запуске поиска, поэтому небезопасно использовать один и тот же запрос одновременно в нескольких потоках. Если код создан с использованием
RDK_BUILD_THREADSAFE_SSSаргумента (по умолчанию для предоставляемых нами двоичных файлов), используется мьютекс, чтобы гарантировать, что только один поток использует данный рекурсивный запрос в каждый момент времени.Вызов MolToSmiles() для одной и той же молекулы из нескольких потоков может привести к гонкам данных с вычисляемыми свойствами молекулы.
Реализация дескриптора TPSA ¶
Дескриптор топологической полярной поверхности (TPSA), реализованный в RDKit, описан в публикации Питера Эртла и др. ( https://pubs.acs.org/doi/abs/10.1021/jm000942e ) Реализация RDKit отличается от того, что описано в этой публикации. В этом разделе описывается разница и почему она есть.
Реализация TPSA в RDKit по умолчанию включает только вклады атомов N и O. Однако таблица 1 публикации TPSA включает параметры для полярных S и P в дополнение к N и O. Что происходит?
Оригинальная реализация TPSA, которая находится в каталоге Daylight Contrib ( http://www.daylight.com/download/contrib/tpsa.html ), не включает вклады от полярных S или P, и, как оказалось, справочные значения, включенные в статью TPSA, также не включают вклады S или P. Например, TPSA, представленная в таблице 3 для фоскарнета (SMILES OC(=O)P(=O)(O)O ), 94,8, соответствует сумме вкладов O - 3x20,23 + 2*17,07 = 94,8 . Добавление вклада P - 9,81 - даст PSA 104,6. Это также верно для других соединений, содержащих P и S, в таблице 3.
В реализации RDKit мы решили воспроизвести поведение программы tpsa.c Contrib и то, что указано в Таблице 3 статьи, поэтому полярные S и P игнорируются. На основании нескольких запросов пользователей в выпуске RDKit 2018.09 мы добавили возможность включать вклады S и P:
>>> from rdkit.Chem import Descriptors
>>> Descriptors.TPSA(Chem.MolFromSmiles('OC(=O)P(=O)(O)O')) # foscarnet
94.83
>>> Descriptors.TPSA(Chem.MolFromSmiles('OC(=O)P(=O)(O)O'), includeSandP=True)
104.64...
>>> Descriptors.TPSA(Chem.MolFromSmiles('Cc1ccccc1N1C(=O)c2cc(S(N)(=O)=O)c(Cl)cc2NC1C')) # metolazone
92.5
>>> Descriptors.TPSA(Chem.MolFromSmiles('Cc1ccccc1N1C(=O)c2cc(S(N)(=O)=O)c(Cl)cc2NC1C'), includeSandP=True)
100.88
Свойства Atom и файлы SDF ¶
Примечание. В этом разделе описываются функциональные возможности, добавленные в версии RDKit 2019.03.1 .
По умолчанию классы rdkit.Chem.rdmolfiles.SDMolSupplierи rdkit.Chem.rdmolfiles.ForwardSDMolSupplier( RDKit::SDMolSupplierи RDKit::ForwardMolSupplierв C++) теперь могут распознавать некоторые молекулярные свойства как списки свойств и преобразовывать их в атомарные свойства. Свойства с именами, начинающимися с atom.prop, atom.iprop, atom.dprop, или atom.bprop
преобразуются в атомарные свойства типа string, int (64 бит), double или bool соответственно.
Вот пример блока из SDF, демонстрирующий все функции, они описаны ниже:
property_example
RDKit 2D
3 3 0 0 0 0 0 0 0 0999 V2000
0.8660 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.4330 0.7500 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
-0.4330 -0.7500 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0
2 3 1 0
3 1 1 0
M END
> <atom.dprop.PartialCharge> (1)
0.008 -0.314 0.008
> <atom.iprop.NumHeavyNeighbors> (1)
2 2 2
> <atom.prop.AtomLabel> (1)
C1 N2 C3
> <atom.bprop.IsCarbon> (1)
1 0 1
> <atom.prop.PartiallyMissing> (1)
one n/a three
> <atom.iprop.PartiallyMissingInt> (1)
[?] 2 2 ?
$$$$
Каждый список свойств атома должен содержать число разделенных пробелами элементов, равное числу атомов. Отсутствующие значения по умолчанию обозначаются строкой n/a. Маркер отсутствующего значения можно изменить, начав список свойств со значения в квадратных скобках. Так, например, свойство PartiallyMissingустанавливается на «один» для атома 0, на «три» для атома 2 и не устанавливается для атома 1. Аналогично свойство PartiallyMissingIntустанавливается на 2 для атома 0, на 2 для атома 1 и не устанавливается для атома 2.
Такое поведение включено по умолчанию и может быть включено/выключено с помощью
rdkit.Chem.rdmolfiles.SetProcessPropertyListsметода.
Если у вас есть свойства атома, которые вы хотели бы записать в файлы SDF, вы можете использовать функции
rdkit.Chem.rdmolfiles.CreateAtomStringPropertyList(), rdkit.Chem.rdmolfiles.CreateAtomIntPropertyList(),
rdkit.Chem.rdmolfiles.CreateAtomDoublePropertyList(), или rdkit.Chem.rdmolfiles.CreateAtomBoolPropertyList():
>>> m = Chem.MolFromSmiles('CO')
>>> m.GetAtomWithIdx(0).SetDoubleProp('foo',3.14)
>>> Chem.CreateAtomDoublePropertyList(m,'foo')
>>> m.GetProp('atom.dprop.foo')
'3.1400000000000001 n/a'
>>> from io import StringIO
>>> sio = StringIO()
>>> w = Chem.SDWriter(sio)
>>> w.write(m)
>>> w=None
>>> print(sio.getvalue())
RDKit 2D
2 1 0 0 0 0 0 0 0 0999 V2000
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.2990 0.7500 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0
M END
> <atom.dprop.foo> (1)
3.1400000000000001 n/a
$$$$
Поддержка улучшенной стереохимии ¶
Обзор ¶
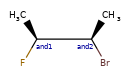
Расширенная стереохимия используется для указания того, что молекула представляет собой более одного возможного диастереомера.
ANDуказывает, что молекула представляет собой смесь молекул. ORуказывает на неизвестные отдельные вещества, а ABSуказывает на отдельное вещество. Это следует соглашению, используемому в файлах V3k mol: группы атомов с указанной стереохимией с маркером ABS, AND, или OR, указывающим на то, что известно.
Вот несколько иллюстраций того, что означают различные комбинации:
Что нарисовано |
Смесь? |
Что это значит |
|---|---|---|
|
смесь |
|
|
смесь |
|
|
смесь |
|
|
одинокий |
|
|
одинокий |
|
|
одинокий |
|
|
смесь |
|
|
одинокий |
|
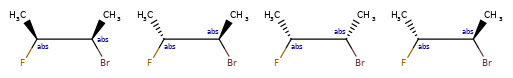
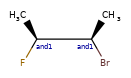
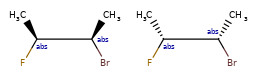
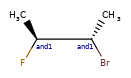
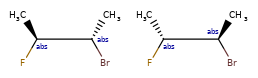
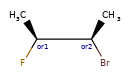
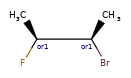
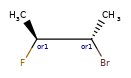
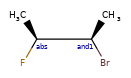
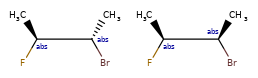
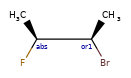
Представление ¶
Хранится как вектор rdkit.Chem.rdchem.StereoGroupобъектов на молекуле. Каждый StereoGroupотслеживает свой тип и набор атомов, которые его составляют.
Варианты использования ¶
Первоначальная цель — не терять данные при передаче туда и обратно. Манипулирование и изображение — это будущие цели.V3k mol -> RDKit -> V3k mol
Можно перечислить элементы a, StereoGroupиспользуя функцию
rdkit.Chem.EnumerateStereoisomers.EumerateStereoisomers(). Обратите внимание, что это удаляет StereoGroupинформацию из продуктов, поскольку теперь они соответствуют определенным молекулам:
>>> m = Chem.MolFromSmiles('C[C@H](F)C[C@H](O)Cl |a:4,&1:1|')
>>> m.GetStereoGroups()[0].GetGroupType()
rdkit.Chem.rdchem.StereoGroupType.STEREO_ABSOLUTE
>>> [x.GetIdx() for x in m.GetStereoGroups()[0].GetAtoms()]
[4]
>>> m.GetStereoGroups()[1].GetGroupType()
rdkit.Chem.rdchem.StereoGroupType.STEREO_AND
>>> [x.GetIdx() for x in m.GetStereoGroups()[1].GetAtoms()]
[1]
>>> from rdkit.Chem.EnumerateStereoisomers import EnumerateStereoisomers
>>> [Chem.MolToCXSmiles(x) for x in EnumerateStereoisomers(m)]
['C[C@@H](F)C[C@H](O)Cl', 'C[C@H](F)C[C@H](O)Cl']
Реакции также сохраняются до тех пор, пока реакция не создает или не разрушает хиральность атома.StereoGroup``s. Product atoms are included in the ``StereoGroup
>>> def clearAllAtomProps(mol):
... """So that atom mapping isn't shown"""
... for atom in mol.GetAtoms():
... for key in atom.GetPropsAsDict():
... atom.ClearProp(key)
...
>>> rxn = AllChem.ReactionFromSmarts('[C:1]F >> [C:1]Br')
>>> ps=rxn.RunReactants([m])
>>> clearAllAtomProps(ps[0][0])
>>> Chem.MolToCXSmiles(ps[0][0])
'C[C@H](Br)C[C@H](O)Cl |a:4,&1:1|'
Stereo Groups can be canonicalized.
>>> m = Chem.MolFromSmiles('CC(C)[C@H]1CCCCN1C(=O)[C@H]1CC[C@@H](C)CC1 |a:3,o1:11,o2:14|')
>>> mOut = Chem.CanonicalizeStereoGroups(m, Chem.StereoGroupAbsOptions.NeverInclude)
>>> Chem.MolToCXSmiles(mOut)
'CC(C)[C@H]1CCCCN1C(=O)[C@H]1CC[C@H](C)CC1 |o1:14|'
>>> mOut = Chem.CanonicalizeStereoGroups(m, Chem.StereoGroupAbsOptions.AlwaysInclude)
>>> Chem.MolToCXSmiles(mOut)
'CC(C)[C@H]1CCCCN1C(=O)[C@H]1CC[C@H](C)CC1 |a:3,11,o1:14|'
>>> mOut = Chem.CanonicalizeStereoGroups(m, Chem.StereoGroupAbsOptions.OnlyIncludeWhenOtherGroupsExist)
>>> Chem.MolToCXSmiles(mOut)
'CC(C)[C@H]1CCCCN1C(=O)[C@H]1CC[C@H](C)CC1 |a:3,11,o1:14|'
>>> m = Chem.MolFromSmiles('CC(C)[C@H]1CCCCN1C(=O)[C@H]1CC[C@@H](C)CC1 |a:3|')
>>> mOut = Chem.CanonicalizeStereoGroups(m, Chem.StereoGroupAbsOptions.OnlyIncludeWhenOtherGroupsExist)
>>> Chem.MolToCXSmiles(mOut)
'CC(C)[C@H]1CCCCN1C(=O)[C@H]1CC[C@@H](C)CC1'
Расширенный поиск стереохимии и субструктуры ¶
Расширенная стереохимия может быть факультативно учтена в поиске подструктур. Следующая таблица фиксирует, соответствует ли запрос подструктуры (в строках) определенной молекуле (в столбцах).
|
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|---|
|
И |
И |
И |
И |
И |
И |
И |
|
Н |
И |
Н |
Н |
Н |
И |
И |
|
Н |
Н |
И |
Н |
Н |
И |
И |
|
Н |
Н |
Н |
И |
Н |
Н |
Н |
|
Н |
И |
Н |
Н |
И |
И |
И |
|
Н |
Н |
Н |
Н |
Н |
И |
И |
|
Н |
Н |
Н |
Н |
Н |
Н |
И |
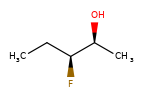
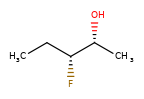
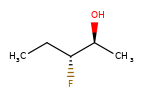
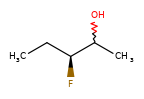
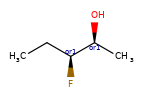
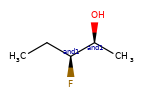
Поиск субструктуры с использованием молекул с улучшенной стереохимией следует следующим правилам (где субструктура < суперструктура):
ахиральный < все, потому что ахиральный запрос означает игнорировать хиральность в сопоставлении
хиральный < И, потому что И включает в себя как хиральную молекулу, так и другую
хиральный < OR, потому что OR включает либо хиральную молекулу, либо другую
ИЛИ < И, потому что И включает обе молекулы, которые ИЛИ может фактически означать.
одна группа из двух атомов < двух групп из одного атома, поскольку последняя представляет собой 4 различных диастереомера, а первая — только два из четырех.
Вот несколько конкретных примеров:
>>> ps = Chem.SubstructMatchParameters()
>>> ps.useChirality = True
>>> ps.useEnhancedStereo = True
>>> m_ABS = Chem.MolFromSmiles('CC[C@H](F)[C@H](C)O')
>>> m_AND = Chem.MolFromSmiles('CC[C@H](F)[C@H](C)O |&1:2,4|')
>>> m_OR = Chem.MolFromSmiles('CC[C@H](F)[C@H](C)O |o1:2,4|')
>>> m_AND.HasSubstructMatch(m_ABS,ps)
True
>>> m_OR.HasSubstructMatch(m_ABS,ps)
True
>>> m_AND.HasSubstructMatch(m_OR,ps)
True
>>> m_OR.HasSubstructMatch(m_AND,ps)
False
Атропоизомерные связи ¶
Некоторые одинарные связи имеют ограниченное вращение из-за стерических взаимодействий между группами на соседних атомах. Если группы на соседних атомах отличаются друг от друга, может быть вызвана хиральность. Атропоизомерная связь является такой ограниченной вращательной связью.
Требования к связи, подлежащей атропоизомеризации в RDKit, следующие:
Связь должна быть одинарной между гибридизированными атомами SP2.
Соседние связи должны быть одинарными, двойными или ароматическими.
Если на каждом конце имеется две группы, то эти группы должны быть разными в соответствии с правилами CIP.
В настоящее время RDKit рассматривает кольцевые связи как потенциальные атропоизомерные связи только в том случае, если кольцо, в котором появляется связь, состоит из 8 атомов или более (что допускает макроциклы).
Для интерпретации атропоизомерных связей молекула должна иметь координаты.
Определение потенциальных атропоизомерных связей основано на расклинивании соседних связей.
Определение атропоизомеров ¶
По крайней мере одна из соседних связей одного из атомов потенциальной атропоизомерной связи должна быть одинарной или ароматической связью и должна иметь направление связи, которое либо «клиновидное», либо «хешированное». Если какая-либо из соседних связей помечена как «сквиггли», считается, что связь имеет «любую» стереохимию. Пример структуры:
Если более одной из соседних связей заклинены или хешированы, они должны быть последовательны.
Например, если две соседние связи на одном и том же конечном атоме заклинены/хешированы, одна должна быть клином, а другая должна быть хешем. Если соседние связи на разных концах связи атропоизомера заклинены, и две связи находятся на противоположных сторонах потенциальной связи атропоизомера (двугранный угол больше 90 градусов или меньше -90 градусов), обе должны быть клиньями или обе должны быть хешированы. Если две заклиненные/хешированные соседние связи находятся на одной стороне потенциальной связи атропоизомера (двугранный угол меньше 90 градусов и больше -90 градусов), одна должна быть клином, а другая хешем.
Примеры – действительные атропоизомеры с несколькими клиньями:
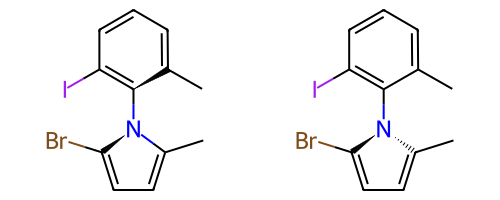
Примеры – недействительные атропоизомеры с несколькими клиньями:
Примечание: программное обеспечение RDKit не пытается определить, действительно ли связь ограничена вращением. Если связь соответствует указанным выше требованиям, она помечается как атропоизомер.
Внутреннее представление атропоизомеров ¶
Чтобы облегчить объяснение, мы включаем индексы связей в рисунок молекулы:
Вот часть отладочного вывода для этой молекулы:
Atoms:
...
1 6 C chg: 0 deg: 3 exp: 4 imp: 0 hyb: SP2 arom?: 1
...
5 6 C chg: 0 deg: 3 exp: 4 imp: 0 hyb: SP2 arom?: 1
...
7 6 C chg: 0 deg: 3 exp: 4 imp: 0 hyb: SP2 arom?: 1
8 7 N chg: 0 deg: 3 exp: 3 imp: 0 hyb: SP2 arom?: 1
9 6 C chg: 0 deg: 3 exp: 4 imp: 0 hyb: SP2 arom?: 1
...
13 6 C chg: 0 deg: 3 exp: 4 imp: 0 hyb: SP2 arom?: 1
...
Bonds:
...
6 5->7 order: a conj?: 1 aromatic?: 1
7 7->8 order: 1 stereo: CCW bonds: (14 6 8 15) conj?: 1
8 8->9 order: a conj?: 1 aromatic?: 1
...
14 7->1 order: a dir: wedge conj?: 1 aromatic?: 1
15 13->8 order: a conj?: 1 aromatic?: 1
Это говорит нам о том, что связь 7 является атропоизомерной и что при взгляде сверху вниз по связи (от атома C7 к атому N8) направление вращения между связями 14 и 8 (это связи с атомами с наименьшим номером в каждой атропоизомерной связи) — против часовой стрелки (CCW).
Форматы, поддерживающие атропоизомеры ¶
Атропоизомеры поддерживаются для анализа молекул и записи в форматах Mol, MRV и CXSmiles. Для реакций атропоизомеры поддерживаются в форматах rxn, MRV и CXSmiles. Атропоизомеры могут быть проанализированы из файла CDXML.
Улучшенная стереохимия ¶
Атропоизомеры могут быть частью расширенных стереохимических групп (Или, И или Абсолютные). Это указывается путем маркировки одного или обоих атомов связи атропоизомера как находящихся в расширенной стереогруппе
Пример:
3D-координаты для ввода атропоизомеров ¶
Если доступны 3D-координаты (а 2D-координаты отсутствуют), связь атропоизомера маркируется только в том случае, если одна из соседних связей заклинена/хеширована. Информация о заклинивании/хешировании игнорируется, за исключением сигнализации о наличии атропоизомера. Фактическая конфигурация определяется 3D-координатами.
Вот пример. Рисунок слева показывает 3D-структуру с клиновидной связью на одном конце связи атропоизомера. Рисунок справа показывает 2D-чертеж той же структуры. В обоих случаях связь атропоизомера выделена красным. Обратите внимание, что клиновидность связи в 3D-структуре не соответствует 3D-координатам; она просто используется для указания того, что есть связь атропоизомера, фактическая стереохимия связи определяется 3D-координатами.
Интерпретация атропоизомеров без координат ¶
Так же, как можно интерпретировать атомную и двойную стереохимию связей из SMILES без указания атомных координат, RDKit принимает (начиная с версии 2024.03.2) соглашение для интерпретации информации о клиньях связей в CXSMILES, для которой не указаны координаты.
Чтобы понять, как работает это представление, необходимо сделать небольшое отступление, чтобы объяснить разницу в нумерации связей в CXSMILES и RDKit. В RDKit связи замыкания кольца добавляются последними; они имеют наивысший номер в молекуле. В CXSMILES связи замыкания кольца добавляются сразу после замыкания кольца. Вот иллюстрация этого для SMILES CC1=CC=CC(I)=C1N1C(C)=CC=C1Br; нумерация связей RDKit (то, что мы видели выше) показана первой, а нумерация CXSMILES — второй.
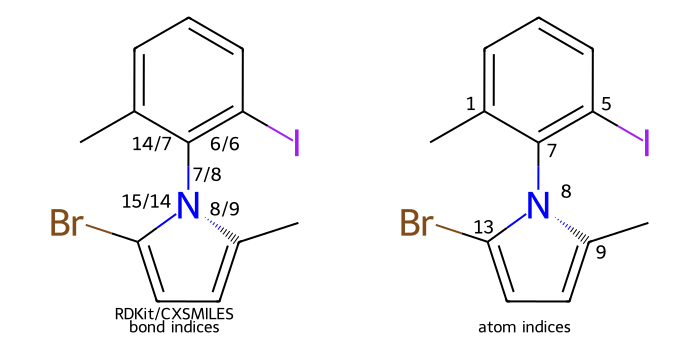
Чтобы описать этот атропоизомер в CXSMILES без использования координат, мы принимаем соглашение, что стереохимия атропоизомерной связи такова, CCWчто связь с соседом с наименьшим номером начального атома (в данном случае сосед, соединенный через связь 7) заклинена, и записываем CXSMILES для этой молекулы как . Стереозначения для всех возможных комбинаций показаны здесь:CC1=CC=CC(I)=C1N1C(C)=CC=C1Br |wU:7.7||
от |
к |
индекс облигаций |
тип |
стерео |
|
|---|---|---|---|---|---|
7 |
1 |
7 |
клин |
CCW |
|
7 |
5 |
6 |
клин |
CW |
|
8 |
9 |
9 |
клин |
CW |
|
8 |
13 |
14 |
клин |
CCW |
|
7 |
1 |
7 |
бросаться |
CW |
|
7 |
5 |
6 |
бросаться |
CCW |
|
8 |
9 |
9 |
бросаться |
CCW |
|
8 |
13 |
14 |
бросаться |
CW |
|
Также возможно вклинивание нескольких соседних связей вокруг атропоизомерной связи, но вклинивание должно быть последовательным в соответствии с таблицей выше; поэтому CXSMILES является допустимым, но не является таковым.CC1=CC=CC(I)=C1N1C(C)=CC=C1Br |wD:7.7,wU:8.9|CC1=CC=CC(I)=C1N1C(C)=CC=C1Br |wD:7.7,wU:8.14|
Проверка атропоизомеров ¶
Недействительные атропоизомеры (например, имеющие две эквивалентные группы на одном конце) будут удалены при считывании молекул с включенной очисткой или при вызове cleanupAtropisomers(mol).
Поиск и канонизация ¶
Атропоизомеры поддерживаются в алгоритме канонизации RDKit. В настоящее время они игнорируются при поиске подструктур и поиске подобия.
Функции запроса в рисунках молекул ¶
Компактное и четкое включение информации о функциях запроса в рисунки молекул является сложной задачей. Это, безусловно, работа в процессе, но в этом разделе описывается то, что поддерживается в настоящее время.
Запрос облигаций ¶
Ниже приведен пример изображения, показывающего, как отображаются различные типы облигаций и запросов-облигаций.
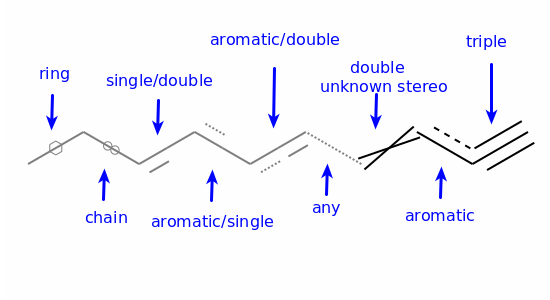
Здесь явно есть место для улучшения, например, не так-то просто отличить облигации «Любые» от запросов, где не реализована специальная обработка («другие» типы запросов):
Запрос атомов ¶
На данный момент единственной реальной поддержкой функций атомарных запросов является рендеринг списков атомов (и списков «НЕ» атомов); другие атомарные запросы рендерятся с помощью простого ? :
Поколение конформистов ¶
Введение ¶
RDKit может генерировать конформеры для молекул, используя два разных метода. Оригинальный метод использовал геометрию расстояний. [ 16 ] По умолчанию используется следующий алгоритм:
Матрица расстояний молекулы рассчитывается на основе таблицы связей и набора правил.
Матрица границ сглаживается с помощью алгоритма сглаживания границ треугольников.
Генерируется случайная матрица расстояний, удовлетворяющая матрице границ.
Эта матрица расстояний встроена в трехмерные измерения (создавая координаты для каждого атома).
Полученные координаты несколько очищаются с помощью «силового поля геометрии расстояний» на основе ограничений расстояния из матрицы границ.
RDKit также имеет реализацию метода ETKDG Риникера и Ландрума [ 17 ] , который изменяет шаг 5 выше, чтобы также использовать предпочтения угла кручения из Кембриджской структурной базы данных (CSD) для исправления конформеров после того, как для их генерации использовалась геометрия расстояний. Подход ETDKDG можно расширить, включив дополнительные термины кручения для малых колец и/или макроциклов [ 18 ] .
При использовании подходов ETKDG качество генерируемых конформеров, как правило, достаточно хорошее, чтобы позволить использовать их «как есть» (т.е. без последующего этапа минимизации с другим силовым полем) для многих приложений.
Параметры, контролирующие генерацию конформеров ¶
В классе доступно большое количество параметров, которые позволяют контролировать процесс генерации конформера EmbedParameters. Подмножество особенно полезных параметров описано здесь:
randomSeed: (по умолчанию -1) позволяет задать случайное начальное число, чтобы обеспечить воспроизводимые результаты.numThreads: (по умолчанию 1) устанавливает количество вычислительных потоков, используемых при генерации нескольких конформеров. Если установлено значение 0, будет использоваться максимальное количество потоков, разрешенное в вашей системе.useRandomCoords: (по умолчанию False) если установлено значение True, то будет выполнено встраивание случайных координат: вместо шагов 3 и 4 выше, атомы будут случайным образом помещены в ящик, а затем их положения будут минимизированы с помощью «силового поля геометрии расстояния» на шаге 5. Этот подход был описан в ссылке [ 19 ]enforceChirality: (по умолчанию True) гарантирует, что хиральность указанных стереоцентров в молекуле сохраняется в конформерах.embedFragsSeparately: (по умолчанию True) для молекул, состоящих из нескольких разъединенных фрагментов, это приводит к тому, что конформеры фрагментов генерируются независимо друг от друга.coordMap: (по умолчанию пусто) можно использовать для предоставления трехмерных координат, которые будут использоваться для ограничения положений некоторых атомов в молекуле.boundsMat: (по умолчанию пусто) может использоваться для предоставления матрицы границ расстояний для молекулы.useExpTorsionAnglePrefs: (по умолчанию False) использовать часть ET ETKDG [ 17 ]useBasicKnowledge: (по умолчанию False) использовать часть K ETKDG [ 17 ]ETVersion: (по умолчанию 1) указать версию стандартных определений кручения для использования. ПРИМЕЧАНИЕ для ETKDGv2 и ETKDGv3 это должно быть 2, поскольку ETKDGv3 использует определения ETKDGv2 для стандартных кручений (извините за запутанную нумерацию)useSmallRingTorsions: (по умолчанию False) использовать часть sr из srETDKGv3 [ 18 ]useMacrocycleTorsions: (по умолчанию False) использовать макроциклические торсионы из ETKDGv3 [ 18 ]useMacrocycle14config: (по умолчанию False) использовать границы расстояния 1-4 из ETKDGv3 [ 18 ]forceTransAmides: (по умолчанию True) ограничить амидные связи транс-конфигурациейpruneRMsThresh: (по умолчанию -1,0) если >0,0, включается обрезка RMSD конформеровonlyHeavyAtomsForRMS: (по умолчанию: False) включает игнорирование атомов H при выполнении обрезки RMSDuseSymmetryForPruning: (по умолчанию True) использует симметрию для вычисления минимального RMSD между двумя конформерами при выполнении обрезки RMSD. Обратите внимание, что включение этого параметра заставляет вычисление RMSD действовать так, как будто onlyHeavyAtomsForRMS установлено в true (даже если сам параметр установлен в False).
Обратите внимание, что существуют предварительно настроенные объекты параметров для доступных версий ETKDG: ETKDG, ETKDGv2, ETKDGv3, иsrETKDGv3
Дополнительная информация об ДÅктიЛიскооРи ¶
Этот раздел, который в настоящее время не является исчерпывающим, призван предоставить некоторую документацию о типах ДÅктიЛიскооРи, доступных в RDKit. Мы не воспроизводим информацию, которую можно найти в литературе, но пытаемся захватить неопубликованные части (которых довольно много).
ДÅктიЛიскооРи RDKit ¶
Это специфичный для RDKit ДÅктიЛიскооРи, который вдохновлен (хотя и существенно отличается от) общедоступными описаниями отпечатка ДÅктიЛიскооРи Daylight [ 7 ] . Алгоритм отпечатка ДÅктიЛიскооРи определяет все подграфы в молекуле в пределах определенного диапазона размеров, хэширует каждый подграф для генерации идентификатора сырого бита, модифицирует этот идентификатор сырого бита для соответствия назначенному размеру отпечатка ДÅктიЛიскооРи, а затем устанавливает соответствующий бит. Доступны опции для генерации основанных на подсчете форм отпечатка ДÅктიЛიскооРи или «несложенных» форм (с использованием разреженного представления).
- Стандартная схема хеширования подграфов заключается в хешировании отдельных связей на основе:
типы двух атомов. Типы атомов включают атомный номер (mod 128) и является ли атом ароматическим.
степени двух атомов на пути.
тип связи (или
AROMATICесли связь обозначена как ароматическая)
Параметры, специфичные для ДÅктიЛიскооРи ¶
minPathиmaxPathконтролировать размер (в связях) рассматриваемых подграфов/путей
nBitsPerHash: Если это больше единицы, каждый подграф установит более одного бита. Дополнительные биты будут сгенерированы путем заполнения генератора случайных чисел исходным необработанным идентификатором бита и генерации соответствующего количества случайных чисел.
useHs: переключает включение или выключение H в подграфы/пути (при условии, что в графе молекул есть H).
tgtDensity: если это больше нуля, отпечаток ДÅктიЛიскооРи будет многократно сложен пополам до тех пор, пока плотность установленных битов не станет больше или равной этому значению или отпечаток ДÅктიЛიскооРи будет содержать только биты minSize . Обратите внимание, что это означает, что полученный отпечаток ДÅктიЛიскооРи не обязательно будет иметь запрошенный вами размер.
branchedPaths: если это значение true (значение по умолчанию), алгоритм будет использовать подграфы (т. е. объекты могут быть разветвленными). Если false, будут рассматриваться только линейные пути.
useBondOrder: если true (по умолчанию), типы связей будут учитываться при хешировании подграфов, в противном случае этот компонент хэша будет игнорироваться.
Образец ДÅктიЛიскооРи ¶
Эти отпечатки ДÅктიЛიскооРи были разработаны для использования при скрининге подструктур. Насколько мне известно, они уникальны для RDKit. Алгоритм идентифицирует особенности в молекуле, выполняя поиск подструктур с использованием небольшого числа (12 в выпуске
2019.03RDKit) очень общих шаблонов SMARTS — например,
[*]~[*]~[*](~[*])~[*]или [R]~1[R]~[R]~[R]~1, а затем хэшируя каждое вхождение шаблона на основе задействованных типов атома и связи. Тот факт, что конкретный шаблон вообще совпал с молекулой, также сохраняется путем хэширования идентификатора и размера шаблона. Если конкретный элемент содержит либо атом запроса, либо связь запроса (например, что-то сгенерированное из SMARTS), единственной информацией, которая хэшируется, является факт соответствия общего шаблона.
Для 2019.03высвобождения типы атомов используют только атомный номер атома, а типы связей используют тип связи или ( AROMATICдля ароматических связей)).
ПРИМЕЧАНИЕ : Поскольку он играет важную роль в отслеживании подструктуры, внутренние элементы этого отпечатка ДÅктიЛიскооРи (используемые общие шаблоны и/или детали алгоритма хеширования) могут меняться от версии к версии.
Атомно-парные и топологические торсионные ДÅктიЛიскооРи ¶
Эти два связанных отпечатка ДÅктიЛიскооРи реализованы на основе оригинальных статей: [ 8 ] [ 9 ] . Атомы типизируются на основе атомного номера, числа пи-электронов и степени атома. При желании информация об атомной хиральности также может быть интегрирована в типы атомов. Оба типа отпечатков ДÅктიЛიскооРи могут быть сгенерированы в явной или разреженной форме и как битовые или счетные векторы. Эти типы отпечатков ДÅктიЛიскооРи отличаются от других в RDKit тем, что биты в разреженной форме отпечатка ДÅктიЛიскооРи могут быть напрямую объяснены (т. е. используемая «функция хеширования» полностью обратима).
Эти отпечатки ДÅктიЛიскооРи изначально «предназначались» для использования в векторах подсчета, и, похоже, так они работают лучше. Поведение по умолчанию явных форм бит-векторов обоих отпечатков ДÅктიЛიскооРи заключается в использовании процедуры «симуляции подсчета», когда для заданного признака устанавливается несколько битов, если он встречается более одного раза. Поведение по умолчанию заключается в использовании 4 битов отпечатка ДÅктიЛიскооРи для каждого признака (поэтому 2048-битный отпечаток ДÅктიЛიскооРи фактически хранит информацию о том же количестве признаков, что и 512-битный отпечаток ДÅктიЛიскооРи, который не использует симуляцию подсчета). Ячейки соответствуют подсчетам 1, 2, 4 и 8. Вот пример того, как это работает: если признак встречается в молекуле 5 раз, будут установлены биты, соответствующие подсчетам 1, 2 и 4.
Морган и особенности МоргОНÅ ДÅктიЛიскооРи ¶
Они реализованы на основе оригинальной статьи [ 10 ] . Алгоритм следует описанию в статье настолько близко, насколько это возможно, за исключением определений химических признаков, используемых для отпечатка ДÅктიЛიскооРи «Feature Morgan» — реализация RDKit использует типы признаков Donor, Acceptor, Aromatic, Halogen, Basic и Acidic с определениями, адаптированными из статьи [ 11 ] . Можно предоставить собственные типы атомов. Отпечатки ДÅктიЛიскооРи доступны либо как явные или разреженные векторы количества, либо как явные битовые векторы.
Многослойные ДÅктიЛიскооРи ¶
Это еще один «оригинал RDKit», и он был разработан с намерением использовать его в качестве подструктурного ДÅктიЛიскооРи. Поскольку шаблонный ДÅктიЛიскооРи гораздо проще и оказался весьма эффективным в качестве подструктурного ДÅктიЛიскооРи, слоистый отпечаток ДÅктიЛიскооРи не получил особого внимания. Он все еще может быть интересен для чего-то, поэтому мы продолжаем включать его.
Идея отпечатка ДÅктიЛიскооРи заключается в генерации признаков с использованием того же алгоритма перечисления подграфов (или путей), который используется в отпечатке ДÅктიЛიскооРи RDKit. После генерации подграфа он используется для установки нескольких битов на основе различных определений типов атомов и связей.
Представления самодостаточной структуры (SCSR) для макромолекул ¶
Поддержка SCSR, добавленная в RDKit, следует описанию в документе BIOVIA «biovia_ctfileformats_2020.pdf», доступном на сайте «CTfile Formats - Dassault Systèmes». Этот документ не содержит никаких реальных подробностей для этого формата и содержит один пример файла.
Кроме того, Biovia Draw поддерживает чтение и запись этого формата. Поддержка RDKit позволяет использовать функциональность, поддерживаемую Biovia Draw, насколько это возможно. Исключением является обработка водородных связей в файлах/блоках SCSR с помощью RDKit (см. ниже).
Представление ¶
Основная моль ¶
Файл SCSR содержит моль с CTAB в формате v3000. Этот CTAB может содержать элементарные атомы и макроатомы, соответствующие аминокислотам (AA), РНК и элементам ДНК. Элементы РНК и ДНК представлены тремя частями – фосфатной группой, сахаром и основанием.
Каждая строка атома в CTAB может ссылаться на элементарный атом или макроатом. Строки макроатома имеют текстовое описание имени макроса и должны иметь атрибуты CLASS и ATTCHORD. Они также могут иметь необязательный атрибут SEQID.
Согласно документу Biovia, атрибут CLASS должен иметь одно из следующих значений: AA, dAA, DNA, RNA, SUGAR, BASE, PHOSPHATE, LINKER, CHEM, LGRP, MODAA, MODdAA, MODDNA, MODRNA, XLINKAA, XLINKdAA, XLINKDNA, XLINKRNA. Для блока SCSR mol (и для любого SGROUP) RDKit требует, чтобы атрибут CLASS имел одно из этих значений.
SEQID — это последовательное целое число, которое игнорируется этой обработкой. Обычно три части элемента РНК или ДНК имеют одинаковый SEQID.
Атрибут ATTCHORD должен иметь спецификацию для каждой связи, которая исходит от макроатома. Спецификация содержится в скобках, а первый символ — это целое число, которое указывает количество последующих элементов. Каждое соединение указывается с помощью идентификатора атома (смещение 1) для связанного атома в основном CTAB и строки, которая указывает точку присоединения для этого макроатома. Строка точки присоединения всегда состоит из двух символов. Первый указывает порядок и представляет собой заглавную букву, начинающуюся с A. Второй символ — один из «l» (левый), «r» (правый), «x» (перекрестные связи — например, для цистеина). В этой реализации второй символ также может быть «h» для водородной связи. Таким образом, присоединённые связи почти всегда находятся в списке: «Al», «Br», «Cx» или «Ch». Например:
ATTCHORD=(6 15 Br 13 Al 21 Cx)
Шаблоны ¶
Заголовок шаблона ¶
В дополнение к CTAB для основной молекулы, каждый макроатом автоматически детализируется в ШАБЛОНЕ. ШАБЛОНЫ пронумерованы и появляются между строками BEGIN TEMPLATES и END TEMPLATES. Каждый шаблон начинается со строки TEMPLATE, которая указывает номер шаблона (от 1 до n), класс и имя шаблона, а также атрибут NATREPLACE.
Класс и имя состоят из трех (или более) частей, разделенных косыми чертами. Первая часть — это КЛАСС, и она должна соответствовать атрибуту КЛАСС в одном или нескольких атомах в основном CTAB. Второй и третий (и последующие) элементы являются вариантами имен шаблона макроатома. Обычно первое имя — это длинная форма, а второе — короткая форма, например «AA/Ala/A» для аланина. Эта обработка не накладывает никаких ограничений на длину имени. Имя, данное макроатому в основном CTAB, должно соответствовать одному из имен в одном из шаблонов с правильным классом. Атрибут NATREPLACE указывает естественную замену для этого макроатома и игнорируется этой обработкой. Пример:
M V30 TEMPLATE 1 SUGAR/Rib/R NATREPLACE=SUGAR/R
Основной шаблон CTAB и SGROUPs ¶
После TEMPLATE идет полный CTAB с атомами и связями шаблона. Каждый CTAB должен содержать SGROUP для основных атомов и связей макроопределения и по одному для каждой уходящей группы. Основная SGROUP для шаблона должна иметь атрибут LABEL, который совпадает с одним из имен в строке TEMPLATE, и он должен иметь атрибут CLASS, который соответствует классу строки TEMPLATE.
Кроме того, основная SGROUP должна иметь атрибуты ATOMS, XBONDS, NATREPLACE. ATOMS — это список всех идентификаторов атомов (смещение 1). XBONDS — это идентификаторы соединительных связей. NATRELACE — это естественная замена, как обсуждалось выше. Атрибуты XBONDS и NATREPLACE игнорируются этой обработкой.
Основная SGROUP также должна содержать список атрибутов SAP (точки присоединения SGROUP). Каждый из них заключен в скобки и начинается с «3». За ними следуют два идентификатора атомов связи, которая начинается в этой SGROUP и идет к атому, НЕ входящему в эту SGROUP, а за этим следует строка, указывающая имя соединения. Имя соединения, как обсуждалось выше, обычно является одним из «Al», «Br», «Cx» или «Ch». SAP «Al», «Br», «Cx» будут иметь один атрибут SAP, а «Ch» обычно будет иметь два или три различных атрибута SAP. Порядок атрибутов SAP для водородных связей («Ch») имеет значение и должен идти от атома водородной связи, ближайшего к соединяющему атому «Al», к самому удаленному. Водородные связи не имеют уходящей группы, поэтому второй идентификатор обозначения — «0».
Пример:
M V30 2 SUP 2 ATOMS=(8 1 2 3 4 5 6 7 8) XBONDS=(1 7)-
M V30 LABEL=U CLASS=BASE SAP=(3 4 9 Al) SAP=(3 8 0 Ch) SAP=(3 6 0 Ch) SAP=(3 7 0 Ch)-
NATREPLACE=BASE/U
В дополнение к основной SGROUP, будет SGROUP, которая идентифицирует каждую уходящую группу. Большинство уходящих групп имеют один атом, но они могут иметь несколько атомов. Уходящие группы SGROUPS имеют атрибуты ATOMS, XBONDS, LABEL и CLASS. Атрибут XBONDS игнорируется в этой обработке. LABEL должен быть «LGRP» (уходящая группа).
Пример:
M V30 1 SUP 1 ATOMS=(1 11) XBONDS=(1 11) LABEL=H CLASS=LGRP
Анализ файлов SCSR и текстовых блоков ¶
RDKit преобразует данные SCSR в полностью атомистическое представление.
SGROUP создается в результирующем RWMol для каждого шаблона и сохраненной уходящей группы из SCSRMol. Имя каждой SGROUP выводится из имени шаблона, порядкового номера, если он присутствует, и, для уходящих групп, идентификатора SAP уходящей группы (например, «Al», «Br», «Cx» или «Ch»).
Следующие вызовы анализируют SCSR mol или block и создают полностью атомистический RDKit mol.:
MolFromSCSRDataStream (поток в mol)
MolFromSCSRBlock (текстовый блок SCSR в mol)
MolFromSCSRFile (файл SCSR в mol)
Все эти функции принимают, в дополнение к имени потока, текстового блока или файла, параметры MolFileParserParams и MolFromSCSRParams. Параметр MolFileParserParams используется для управления разбором данных SCSR, как описано в другом месте этого документа. Параметр MolFromSCSRParams используется для управления преобразованием данных SCSR в полное атомистическое представление.
MolFromScsrParams имеет три свойства:
Свойство «ScsrTemplateNames» управляет тем, как генерируются Sgoups расширенного файла, и должно быть одним из следующих: ScsrTemplateNamesAsEntered,
ScsrTemplateNamesUseFirstName или ScsrTemplateNamesUseLastName. Если указано ScsrTemplateNamesAsEntered, будет использоваться имя, указанное в основном SCSR CTAB.
Свойство «includeLeavingGroups» управляет тем, включаются ли уходящие группы, которые не заменяются в основном CTAB, в результирующий атомистический файл.
Уходящие группы, которые таким образом сохраняются, становятся концевыми крышками и крышками на неиспользуемых сайтах сшивки. Установка этого свойства на «false» приводит к тому, что концевые крышки и крышки сшивки остаются незамещенными. Это позволяет использовать результаты в качестве полного атомного запроса для субъединиц из SCSR mol.
Свойство «SCSRBaseHbondOptions» управляет обработкой водородных связей в файле SCSR. Это описано в следующем разделе.
Водородные связи ¶
Для пар смысл-антисмысл в ДНК и РНК водородные связи представлены как одна водородная связь в представлении SCSR. При преобразовании в полное атомистическое представление каждая такая водородная связь может представлять до 3 водородных связей между полными атомистическими представлениями групп Base.
Обработка водородных связей контролируется членом ScsrBaseHbondOptions параметра ScsrMolFileParserParams. Параметры следующие:
ScsrBaseHbondOptionsIgnore — если этот параметр выбран, все водородные связи игнорируются и не обрабатываются.
ScsrBaseHbondOptionsUseSapAll = 1 Если выбрано, используются все SAP для hbond. Они должны быть определены в шаблоне в правильном порядке, который начинается с первого атома, ближайшего к соединению «Al», и продолжается последовательно
Если на каждой стороне есть 3 сайта, и они комплементарны (доноры соответствуют акцепторам и наоборот), мы добавляем связи. и все готово. Это стандартное связывание Уотсона-Крика (« https://water.lsbu.ac.uk/water/nucleic_acid_hydration.html »).
Если нет, мы проверяем, соответствуют ли они конфигурации связи колебания. Существует четыре общепринятых связи колебания, и мы имеем дело только с этими четырьмя типами. Четыре известных связи колебания:
IC
ИИ
Я
ГУ
«I» обозначает инозин — у него есть только два доступных сайта водородных связей. Пара GU имеет три сайта на каждом конце, но они не являются комплементарными. ( https://en.wikipedia.org/wiki/Wobble_base_pair#:~:text=A%20wobble%20base%20pair%20is,hypoxanthine%2Dcytosine%20(I%2DC ).
Для пар, которые имеют I (инозин) или что-то подобное, конфигурация должна быть DA, поэтому эти два атома образуют две связи H. Другая сторона (C,U или A) имеет конфигурацию AAD (C), ADA (U) или DAD (A). Для оснований типа C и оснований типа A используются вторые и третьи атомы (AD), а для типов U используются первые два атома (AD).
Для пары GU обе стороны имеют по 3 атома, но они не являются комплементарными. Используются второй и третий сайты на стороне G (DA), а первые два сайта на стороне U (AD).
В любом другом случае мы выдвигаем и добавляем только одну связь между первым атомом водородной связи с обеих сторон, даже если они НЕ комплементарны. Это просто указывает на то, что стороны каким-то образом связаны водородной связью и сохраняет общую пару прямой.
ScsrBaseHbondOptionsUseSapOne Если выбрано это, используется только один SAP hbond на базу. Если определено несколько SAP, используется первый сайт hbond, даже если он не лучший. Не делается никаких попыток сопоставить статус донора/акцептора выбранных сайтов связи. (это просто поддерживает связь между парами оснований)
ScsrBaseHbondOptionsAuto Для оснований C,G,A,T,U,In (и производных) используется стандартное водородное связывание Уотсона-Крика, которое определяется путем сопоставления подструктур. В шаблоне не требуется определять водородные связи SAP («Ch»), а если они определены, то игнорируются.
Обработка определенных таким образом участков водородных связей выполняется так же, как и при выборе ScsrBaseHbondOptionsUseSapAll (см. выше).
Пример преобразования пептида:
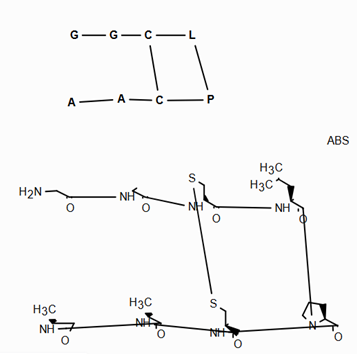
Пример преобразования парных цепей ДНК-РНК:
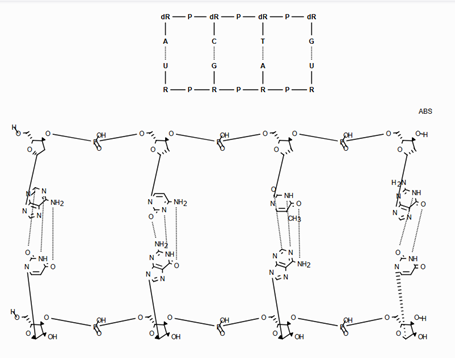
Пример преобразования пары «Колебание»:
Вот два примера файлов SCSR:
ДНКТест3 .
Флаги функций: глобальные переменные, влияющие на поведение RDKit ¶
RDKit использует ряд «флагов функций»: глобальные переменные, которые влияют на его поведение. Они, как правило, добавляются для поддержания обратной совместимости при внедрении новых алгоритмов, которые дают другие результаты.
Вот текущие флаги функций:
preferCoordGen: когда этоtrueбиблиотека Schrodinger's Coordgen с открытым исходным кодом будет использоваться для генерации 2D-координат молекул. Значение по умолчанию —false. Его можно задать из C++ с помощью переменнойRDKit::RDDepict::preferCoordGenили из Python с помощью функцииrdDepictor.SetPreferCoordGen(). Добавлено в выпуске 2018.03.
allowNontetrahedralChirality: когда этоtrueне тетраэдрическая хиральность будет восприниматься из 3D-координат. Значение по умолчанию — ,trueесли переменная окруженияRDK_ENABLE_NONTETRAHEDRAL_STEREOне установлена в"0". Можно задать/проверить из C++ с помощью функцийRDKit::Chirality::setAllowNontetrahedralChirality()/RDKit::Chirality::getAllowNontetrahedralChirality()или из Python с помощью функцийChem.SetAllowNontetrahedralChirality()/Chem.GetAllowNontetrahedralChirality(). Добавлено в выпуске 2022.09.
useLegacyStereoPerception: когда этоtrueустаревшая реализация для восприятия стереохимии, будет использоваться. Значение по умолчанию —trueесли переменная окруженияRDK_USE_LEGACY_STEREO_PERCEPTIONне установлена в"0". Можно установить/проверить из C++ с помощью функцийRDKit::Chirality::setUseLegacyStereoPerception()/RDKit::Chirality::getUseLegacyStereoPerception()или из Python с помощью функцийChem.SetUseLegacyStereoPerception()/Chem.GetUseLegacyStereoPerception(). Добавлено в выпуске 2022.09.
Сноски